作为药物的3,5-双(苯基)-1H-杂芳基衍生物的制作方法
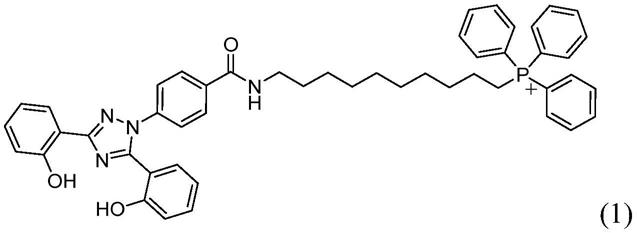
作为药物的3,5-双(苯基)-1h-杂芳基衍生物
技术领域
1.本发明涉及新的3,5-双(苯基)-1h-杂芳基衍生物及其作为药物、特别是用于治疗癌症的用途。
背景技术:
2.癌症是涉及异常细胞生长的疾病。癌症的治疗成本高昂,而且并不总是成功,这是因为癌症通常对标准的化学治疗性治疗相当有抗性。
3.抗癌药物可以通过多种作用机制发挥作用。这些机制包括抗增殖活性、细胞凋亡诱导(或细胞死亡诱导)活性、铁螯合活性和抗迁移活性,以及活性氧(ros)过量产生。
4.所有细胞的dna合成、代谢、生长和增殖都需要铁。铁代表了许多酶促反应所需的不可或缺的微量营养素,诸如核糖核苷酸还原酶对脱氧核糖核苷酸合成的催化活性。它对于线粒体呼吸也是必不可少的,因为它能够接受和献出电子并参与电子传递链,从而导致跨线粒体内膜的电化学梯度的产生。因为癌细胞由于其增殖性质和改变的代谢需求而显示出对铁的需求更高,因此预计铁在肿瘤生长和进展中的重要作用。并且铁代谢的显著改变和肿瘤起始细胞中特定的铁代谢相关基因特征最近得以报道,证明了铁在癌症干细胞样细胞中的重要作用(rychtarcikova等人(2017)tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism.oncotarget 8,6376-6398)。
5.重要的是,癌细胞似乎对产生ros的化合物更敏感,这是因为它们自然升高的基础ros水平和几乎饱和的抗氧化防御机制。因此,ros的任何增加对于增加其抗氧化防御能力非常有限的癌细胞都是有害的,并且这种方法已被用于设计抗癌疗法(kim等人(2016).ros homeostasis and metabolism:a critical liaison for cancer therapy.exp mol med 48,e269;truksa等人(2015).mitochondrially targeted vitamin e succinate modulates expression of mitochondrial dna transcripts and mitochondrial biogenesis.antioxid redox signal 22,883-900)。
6.此外,由于线粒体表示产生血红素和fes簇以用于所有需要它们作为辅助因子的蛋白质的关键细胞器(paul和lill(2015).biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability.biochim biophys acta 1853,1528-1539),对线粒体机制的任何干扰应该导致代谢活动、呼吸和修复dna的能力下降,从而导致细胞周期停滞,不能增殖,即使细胞不发生细胞凋亡并存活。
7.此外,经由称为线粒体自噬的过程,铁和线粒体循环之间存在联系,并与适应性免疫系统的激活相关联,这表明铁处理和线粒体自噬可以改变致癌过程(ziegler等人(2018).mitophagy in intestinal epithelial cells triggers adaptive immunity during tumorigenesis.cell 174,88-101e116)。
8.最近,novartis推出了一种新的螯合剂,称为地拉罗司(dfx,以exjade、desirox、defrijet、desifer、rasiroxpine和jadenu销售)并于2005年在美国获得fda批准(tyagi等
人(2005).deferasirox(icl670;exjade(r)),a novel orally available iron chelator for correction of transfusion-associated iron overload.support cancer ther 2,208-211)。dfx已被证明具有抗癌活性,并且这已在体外以及体内得到证实(tury等人(2018).the iron chelator deferasirox synergises with chemotherapy to treat triple-negative breast cancers.j pathol 246,103-114)。
9.本发明的目标是提供在非常低的水平(nm)下具有细胞抑制作用和在较高浓度(μm)下具有细胞毒性作用而没有显示出不希望的全身作用的化合物。化合物的作用应基于多模式作用机制。
技术实现要素:
10.本发明提供了通式i的新的化合物,
[0011][0012]
其中
[0013]
z是选自亚烷基、亚烯基和亚炔基的线性烃基链,其含有2至20个碳原子、优选6至16个碳原子、更优选6至14个碳原子、甚至更优选8至12个碳原子、最优选10个碳原子或优选8至16个或10至15个碳,其中
[0014]-烃基链中的一个或多个碳原子(通常是-ch
2-基团)可被一个或多个5-元或6-元芳族环或者含有杂原子o、s和/或n的杂芳族环任选替代,所述芳族环或杂芳族环优选选自亚苯基、亚三唑基或亚吡啶基,和/或
[0015]-烃基链中的一个或多个碳原子(通常是-ch
2-基团)可被选自o、s、nh、n-oh的一个或多个杂原子或含杂原子基团任选替代,和/或
[0016]-烃基链原子可为未取代的或被独立选自包括以下的组的一个或多个取代基取代:c1-c4烷基、c1-c4烷氧基、c1-c4烷硫基、c1-c4酰氧基、n(h或c1-c4烷基)2(其中,烷基相同或不同)、苯基、苄基、oh、=o、sh、=s、=n-oh、f、cl、br、i;
[0017]
r1、r2、r3中的每一者独立选自包括以下的组:c1-c10烷基、c6-c12芳基、c6-c12-芳基-c1-c2-烷基、c5-c12杂芳基、c3-c8环烷基,其中,r1、r2、r3中的每一者可以任选地(并独立于其他)被独立选自包括以下的组的一个或多个取代基取代:c1-c4烷基;c1-c4烷氧基;c1-c4烷硫基;n(h或c1-c4烷基)2,其中烷基相同或不同;oh;=o;sh;=s;=n-oh;f;cl;br;i;
[0018]
r4、r5和r6中的每一者选自ch和n,而r4、r5和r6中的至少两者是n;
[0019]
x-是药学上可接受的阴离子;
[0020]
y在每次出现时独立选自包括以下的组:oh、sh、nh2、f、cl、br、i和h。
[0021]
式i的定义旨在包括任何盐、溶剂化物、异构体和异构体混合物。
[0022]
x-通常选自无机酸或有机酸的阴离子,特别合适的是cl-、br-、i-、碳酸根、碳酸氢根、硝酸根、硫酸根、磷酸根、醋酸根、抗坏血酸根、天冬氨酸根、苯磺酸根、苯甲酸根、苯磺酸根、酒石酸氢根、樟脑磺酸根、柠檬酸根、癸酸根、依地酸根、乙磺酸根、甲酸根、富马酸根、葡庚糖酸根、葡糖酸根、谷氨酸根、乙醇酸根、己酸根、羟基萘甲酸根、羟乙基磺酸根、乳酸根、乳糖醛酸根、苹果酸根、马来酸根、扁桃酸根、甲磺酸根、甲基磺酸根、粘液酸根、萘磺酸根、辛酸根、油酸根、草酸根、双羟萘酸根、泛酸根、聚半乳糖醛酸根、丙酸根、水杨酸根、硬脂酸根、琥珀酸根、酒石酸根、茶氯酸根、甲苯磺酸根,但可以使用任何药学上可接受的酸的阴离子。
[0023]
在优选的实施方式中,r6是n,并且r4和r5中的至少一者也是n。
[0024]
在优选的实施方式中,z选自亚烷基;其中一个碳原子被5-元或6-元芳族环或者含有杂原子o、s和/或n的杂芳族环替代的亚烷基;其中一个碳原子被亚苯基、亚三唑基或亚吡啶基替代的亚烷基;其中一个碳原子被nh替代的亚烷基;其中一个碳原子被亚苯基、亚三唑基或亚吡啶基替代、一个碳原子被nh替代并且一个碳原子被=o取代的亚烷基,因此优选形成结构
–
(杂)芳基-c(=o)-nh-。在该优选的实施方式中,z可被任选地进一步取代。
[0025]
在优选的实施方式中,y是h、oh、f或sh。最优选地,y是oh。
[0026]
此外,本发明提供了用作药物的式i化合物,特别用于治疗癌症。癌症可包括例如乳腺癌、卵巢癌、前列腺癌、git癌、肝癌、结直肠癌、胰腺癌、间皮瘤、肺癌和白血病。
[0027]
本发明的化合物通过几种独立的机制起作用,这使得它们适用于治疗各种类型的癌症,包括抗性癌症。式i的化合物的作用机制包括抗增殖活性、诱导细胞凋亡(或诱导细胞死亡)活性、金属螯合活性,特别是铁螯合活性和抗迁移活性。这些活性对恶性细胞具有选择性。抗性癌症通常对通过某种作用机制起作用的药物具有抗性。具有多种作用模式的本发明的化合物则可以通过经由癌细胞对其敏感的不同作用模式起作用来克服抗性。
[0028]
与结构部分相似的物质地拉罗司相比,本发明的代表性化合物显示出对恶性细胞的ic
50
比地拉罗司的ic
50
低3-4个数量级。与另一种结构相似的化合物、他莫昔芬的三苯基癸基鏻衍生物(mitotax,wo2014/173374)相比,本发明的代表性化合物显示出对恶性细胞的ic
50
比mitotax的ic
50
低1-2个数量级。
[0029]
本发明的目标还是药物组合物,其包含至少一种通式i的化合物和至少一种药学上可接受的赋形剂,诸如载体、溶剂、填充剂、粘合剂、崩解剂、包衣、助流剂、润滑剂、保护剂、调味剂、着色剂。
[0030]
另外,本发明包括药学上可接受的组合物,其包含至少一种式i化合物和金属。金属优选选自元素周期表的过渡金属(元素周期表的b族、镧系元素、锕系元素)和iiia族和iva族的金属。金属可以是放射性核素或适合用作诊断剂或治疗剂的金属,例如用于放射疗法、生物传感、生物成像、药物递送、基因递送、光动力疗法(特别是诸如镧、铈、镨、钕、钷、钐、铕、钆、铽、镝、钬、铒、铥、镱、镥、铁、镓、铜的金属)。存在于制剂中的金属总量的至少一部分和式i化合物的总量的至少一部分可以形成螯合络合物。
[0031]
因此,本发明的目的是通式i的化合物或包含至少一种式i的化合物和金属、优选镓的药物制剂作为药物、特别是用于治疗癌症的方法中的用途。
[0032]
本发明的另一目的是通式i的化合物或包含至少一种式i的化合物和金属、优选镓
的药物制剂在制备用于治疗癌症的药物中的用途。
[0033]
此外,本发明的目的是治疗包括人的哺乳动物的方法,其中将一种或多种通式i的化合物施用给患有癌症的对象。
[0034]
本发明的目的还是药物组合物,其包含至少一种式i的化合物和金属、优选镓作为诊断用造影剂、特别是用于体内可视化癌症的方法的用途。此类制剂可以通过例如正电子发射断层扫描(pet)来可视化。
具体实施方式
[0035]
实施例1
[0036]
(10-(4-(3,5-双(2-羟苯基)-1h-1,2,4-三唑-1-基)苯甲酰氨基)癸基)三苯基氯化鏻(化合物1)
[0037]
将地拉罗司(114mg,0.31mmol)与10-氨基癸基氯化鏻盐酸盐(150mg,0.31mmol)、n-(3-二甲基氨基丙基)-n
′‑
乙基碳二亚胺盐酸盐(edc)(88mg,0.46mmol)和n,n-二异丙基乙胺(dipea)(0.32ml,1.86mmol)溶解于二甲基甲酰胺(dmf)(3ml)中,并将反应物搅拌72小时。蒸发溶剂,将混合物溶解于甲醇(meoh)中,并用柠檬酸(5%)、盐水和二氯甲烷(dcm)萃取。作为两种旋转异构体分离的式1的纯产物(25mg,10%)通过柱色谱(2%meoh/chcl3)获得,呈浅黄色泡沫状。
[0038][0039]1h nmr(500mhz,氯仿-d)δ10.96(bs,oh,1h),10.16(bs,oh,1h),8.40(bs,nh,1h),8.12(t,j=7.9hz,2h),7.79
–
7.68(m,6h),7.65(s,7h),7.42(d,j=6.9hz,2h),7.33(t,j=7.9hz,1h),7.25(dd,j=26.3,7.7hz,2h),7.11(d,j=7.7hz,1h),7.00(m,2h),6.74(t,j=7.4hz,1h),3.46(m,4h),1.79
–
1.48(m,6h),1.26(m,10h)。
[0040]
13
c nmr(126mhz,氯仿-d)δ166.48,159.61,157.18,156.65,152.14,139.81,135.84,135.10,133.52(d,j=9.9hz),132.60,131.39,130.52(d,j=12.4hz),129.14,128.79,127.31,124.61,119.65,19.24,118.15(d,j=85.74hz),117.95,117.05,113.65,111.91,40.17,30.19,30.06,29.69,29.00,28.59,28.50,28.46,26.61,22.83,22.51,22.47。
[0041]
c49h50o3n4p
+
hrms计算值:773.36150,实测值:773.36128。
[0042]
实施例2
[0043]
(10-(3,5-双(2-羟苯基)-1h-1,2,4-三唑-1-基)癸基)三苯基溴化鏻(化合物2)
[0044]
将(10-溴癸基)三苯基溴化鏻(750mg,1.334mmol)用肼(6ml)加热2小时。将反应混合物冷却至实验室温度,用h2o(50ml)稀释并用dcm萃取两次并经mgso4干燥。将粗物质浓缩,真空干燥,并用乙醇(6ml)稀释,并加入2-(2-羟苯基)-4h-苯并[e][1,3]噁嗪-4-酮(377mg,
1.576mmol)并将反应混合物回流1.5小时。将反应混合物真空浓缩并通过柱色谱(甲苯:甲醇,梯度100:5
–
100:15)纯化以产生式2的产物(330mg,30%),呈浅黄色泡沫状。
[0045][0046]1h nmr(600mhz,氯仿-d)δ10.55(s,1h),8.06(dd,j=7.8,1.7hz,1h),7.83
–
7.70(m,9h),7.65(td,j=8.0,3.4hz,6h),7.48
–
7.43(m,2h),7.35(ddd,j=8.5,7.2,1.6hz,1h),7.31
–
7.25(m,2h),7.22
–
7.16(m,1h),6.99(ddd,j=7.3,6.5,1.0hz,2h),6.95(td,j=7.5,1.1hz,1h),4.28(t,j=7.4hz,2h),3.64(td,j=12.6,5.1hz,2h),1.96(p,j=7.5hz,2h),1.61
–
1.50(m,4h),1.35
–
1.28(m,2h),1.28
–
1.10(m,8h)。
[0047]
13
c nmr(151mhz,氯仿-d)δ159.10,156.71,156.55,152.15,134.96(d,j=3.0hz),133.58(d,j=10.7hz),132.20,130.85,130.43(d,j=12.5hz),128.23,126.93,119.48,119.40,118.27(d,j=85.8hz),117.97,116.90,114.28,112.61,50.23,30.12(d,j=15.8hz),29.28,28.73(重叠),28.68(d,j=1.1hz),26.18,22.80,22.51,22.48(重叠),22.47。
[0048]
c42h45o2n3p
+
hrms计算值:654.32439,实测值:654.32443。
[0049]
实施例3
[0050]
2,2'-(1-(9-羟壬基)-1h-1,2,4-三唑-3,5-二基)联苯酚(化合物3)
[0051]
将10-溴-1-壬醇(117mg,0.524mmol)与肼(2ml)混合,并在80℃下剧烈搅拌2小时。将反应物冷却至实验室温度,加入水(20ml)并将混合物用dcm(15ml)萃取3次。将粗物质真空浓缩,用乙醇(4ml)稀释,并加入2-(2-羟苯基)-4h-苯并[e][1,3]噁嗪-4-酮(187mg,0.782mmol)。将反应混合物在80℃下加热3小时。将粗产物真空浓缩,并通过色谱柱(氯仿:甲醇,梯度100:0
–
100:1)纯化以产生式3的产物(70mg,24%),呈浅黄色泡沫状。
[0052][0053]1h nmr(600mhz,氯仿-d)δ11.18(s,1h),9.89(s,1h),8.10(dd,j=7.8,1.7hz,1h),7.57(dd,j=7.9,1.6hz,1h),7.46(ddd,j=8.3,7.3,1.6hz,1h),7.37(ddd,j=8.3,7.2,1.7hz,1h),7.19(dd,j=8.4,1.2hz,1h),7.09(dd,j=8.3,1.1hz,1h),7.04(qd,j=7.9,1.1hz,2h),4.47(t,j=7.4hz,2h),3.66(t,j=6.6hz,2h),2.11
–
1.99(m,2h),1.61
–
1.54(m,2h),1.47
–
1.41(m,2h),1.40
–
1.28(m,8h)。
[0054]
13
c nmr(151mhz,氯仿-d)δ158.47,157.54,156.30,151.80,132.53,131.33,127.29,126.67,119.76,119.46,118.33,116.94,113.72,110.93,62.97,50.81,32.65,29.51,29.24,29.16,28.87,26.38,25.60。
[0055]
c23h30o3n3
+
hrms计算值:396.22817,实测值:396.22771。
[0056]
实施例4
[0057]
(10-(3,5-双(2-羟苯基)-1h-1,2,4-三唑-1-基)癸基)三环己基溴化鏻
[0058]
将化合物3(40mg,0.101mmol)溶解于dcm(1ml)中,冷却至0-4℃并一次性加入三苯基二溴化膦(171mg,0.404mmol)。使反应混合物升温至实验室温度并加入dipea(0.2ml)。将混合物在室温下搅拌过夜。将反应物用h2o(0.2ml)猝灭并加入dmf(2ml)。在真空下部分去除h2o和dipea,并加入三环己基膦(1g,3.566mmol)。将反应混合物在真空下在90℃下加热1小时,然后在惰性气氛下继续加热过夜。将反应混合物冷却至实验室温度并将冷却的混合物逐滴加入冷(4℃)二乙醚(200ml)中。形成白色沉淀,倾析出溶剂,并用柱色谱纯化粗产物以产生式4的产物(30mg,40%)。
[0059][0060]1h nmr(600mhz,氯仿-d)δ10.80(s,1h),8.09(dd,j=7.8,1.7hz,1h),7.58
–
7.52(m,1h),7.44(dd,j=7.7,1.7hz,1h),7.42
–
7.32(m,2h),7.30(ddd,j=8.7,7.2,1.7hz,1h),7.02(dd,j=8.3,1.1hz,1h),7.00
–
6.97(m,1h),6.96(dd,j=7.5,1.1hz,1h),4.26(t,j=7.2hz,2h),2.55
–
2.44(m,3h),2.39(dt,j=19.3,6.4hz,2h),2.02
–
1.90(m,8h),1.90
–
1.83(m,6h),1.81
–
1.69(m,4h),1.57
–
1.16(m,27h)。
[0061]
13
c nmr(151mhz,氯仿-d)δ159.32,156.63,156.55,152.31,132.12,130.78,128.52,126.82,119.46,119.34,117.85,116.91,114.37,113.01,49.96,30.68(d,j=13.8hz),29.90(d,j=40.4hz),29.07,28.36(d,j=17.2hz),27.95,27.17,27.14,26.50,26.42,25.76,25.40,22.50(d,j=5.4hz),15.73(d,j=42.6hz)。
[0062]
c41h61o2n3p
+
hrms计算值:658.44959,实测值:658.44945。
[0063]
实施例5
[0064]
4-(2-(叔丁氧基羰基)肼基)苯甲酸(化合物5)
[0065]
将4-肼基苯甲酸(500mg,3.286mmol)和boc酸酐(720mg,3.299mmol,boc=二(叔丁基)二碳酸酯)溶解于dmf(4ml)中,加入dipea(1ml)并将反应混合物搅拌1小时。将反应混合物浓缩并真空浓缩。将粗物质在柱色谱(氯仿:甲醇,100:10)上纯化以得到式5的产物(690mg,79%),呈淡黄色泡沫状。
[0066][0067]1h nmr(600mhz,甲醇-d4)δ7.88
–
7.76(m,2h),6.78
–
6.65(m,2h),1.46(s,6h)。
[0068]
13
c nmr(151mhz,甲醇-d4)δ170.37,158.67,154.93,132.45,121.78,112.01,81.54,28.63。
[0069]
c12h17o4n2
+
hrms计算值:253.11828,实测值:253.11779。
[0070]
实施例6
[0071]
(10-铵基癸基)三苯基氯化鏻盐酸盐(化合物6)
[0072]
将(10-溴癸基)三苯基溴化鏻(9.26g,16.448mmol)溶解于氨在甲醇(60ml)中的7m
溶液,并将反应混合物在50℃下搅拌。6h后,加入另外的在甲醇(40ml)中的氨,然后将反应混合物在50℃下再加热24h。在减压下浓缩反应混合物,并使用硅胶柱色谱(200ml)(氯仿/甲醇梯度0-10%的甲醇)纯化粗产物。将产物用hcl(36%,5ml)酸化并以氯化物形式(50g)通过dowex 1x8过滤,以获得所期望的式6的(10-氨基癸基)三苯基氯化鏻盐酸盐(5.135g,63%)。
[0073][0074]1h nmr(500mhz,甲醇-d4)δ7.92
–
7.85(m,3h),7.85
–
7.68(m,12h),3.46
–
3.37(m,2h),2.90(t,j=7.5hz,2h),1.72
–
1.60(m,4h),1.56(p,j=7.5hz,2h),1.43
–
1.20(m,12h)
[0075]
13
c nmr(126mhz,甲醇-d4)δ136.23(d,j=3.0hz),134.79(d,j=9.9hz),131.51(d,j=12.5hz),120.00(d,j=86.3hz),40.76,31.59(d,j=16.2 5hz),30.29(2c),30.12,29.89,28.53,27.42,23.57(d,j=4.4hz),22.68(d,j=51.1hz)。
[0076]
ir(kbr压片):ν=3051,3007,2927,2854,2006,1825,1601,1587,1485,1465,1438,1402,1337,1318,1189,1161,1113,996,751,723,691。
[0077][0078]
c28h37np
+
hrms计算值:418.26581,实测值:418.26567。
[0079]
实施例7
[0080]
(10-(2,4-双(2-羟苯基)-1h-咪唑-1-基)癸基)三苯基氯化鏻(化合物7)
[0081]
将化合物5(100mg,0.397mmol)、化合物6(389mg,0.793mmol)、o-(苯并三唑-1-基)-n,n,n
′
,n
′‑
四甲基四氟硼酸脲(tbtu)(255mg,0.794mmol)和三乙胺(et3n)(0.55ml,3.9mmol)溶解于dmf(1ml)中,并将反应混合物搅拌2h。将反应混合物滴加至冰冷二乙醚(et2o)(50ml)中,并且油性沉淀在冰浴中搅拌后在烧瓶壁上形成。倾析出溶剂并将沉淀溶解在甲醇(3ml)中并用半饱和nh4cl溶液(10ml)稀释。将所得乳状溶液用dcm(3x 15ml)萃取。将合并的有机层经mgso4干燥并真空浓缩。将相同的萃取程序(3ml甲醇,10ml nh4cl,3x 15mldcm)再重复两次。将粗产物通过柱色谱(氯仿/甲醇梯度0-10%的甲醇)纯化,得到式7的产物(170mg,62%),呈淡黄色泡沫状。
[0082][0083]1h nmr(600mhz,甲醇-d4)δ7.93
–
7.74(m,15h),7.69(d,j=8.7hz,2h),6.81
–
6.75(m,2h),3.44
–
3.37(m,2h),1.67(h,j=8.2hz,2h),1.59(q,j=6.9hz,2h),1.55(t,j=7.5hz,2h),1.51(s,6h),1.33(d,j=24.7hz,12h)。
[0084]
13
c nmr(151mhz,甲醇-d4)δ168.69,157.33,152.31,134.85,133.39(d,j=
10.0hz),130.09(d,j=12.6hz),128.20,124.42,118.88,118.31,110.92,39.39,30.11(d,j=16.1hz),29.14,28.93,28.81(d,j=7.1hz),28.34,27.23,26.54,22.13
–
21.97(m),21.32,20.99。
[0085]
c40h51o3n3p
+
hrms计算值:652.36626,实测值:652.36596。
[0086]
实施例8
[0087]
2-(3,5-双(2-羟苯基)-1h-1,2,4-三唑-1-基)苯甲酸(化合物8)
[0088]
将2-肼基苯甲酸盐酸盐(200mg,1.060mmol)和2-(2-羟苯基)-4h-苯并[e][1,3]噁嗪-4-酮(231mg,0.966mmol)溶解于etoh(10ml)中,并将反应混合物加热至70℃。滴加et3n(0.28ml,2.001mmol)。将反应混合物搅拌4小时,然后加入水(5ml)。蒸发etoh并加入6m hcl(3ml)。粗产物作为浅黄色固体沉淀。用硅胶柱色谱(30ml)(氯仿/甲醇,梯度0-5%的甲醇)获得式8的纯产物(333mg,92%),呈浅黄色泡沫状。
[0089][0090]1h nmr(600mhz,dmso-d6)δ8.00(dd,j=7.8,1.7hz,1h),7.89(dd,j=7.7,1.6hz,1h),7.73(td,j=7.7,1.6hz,1h),7.66
–
7.60(m,2h),7.36(ddd,j=8.3,7.2,1.7hz,1h),7.29(ddd,j=8.3,7.3,1.8hz,1h),7.20(dd,j=7.7,1.7hz,1h),7.02(dd,j=8.3,1.1hz,1h),6.99(td,j=7.5,1.2hz,1h),6.87(dd,j=8.3,1.0hz,1h),6.82(td,j=7.6,1.1hz,1h)。
[0091]
13
c nmr(151mhz,dmso-d6)δ166.28,159.63,156.72,156.20,153.33,136.99,132.57,131.63,131.04(d,j=23.5hz),130.46,128.77,127.10,120.03,119.40,117.41,116.71,114.37,113.95。
[0092]
c21h16o4n3
+
hrms计算值:374.11353,实测值:374.11324。
[0093]
实施例9
[0094]
(10-(2-(3,5-双(2-羟苯基)-1h-1,2,4-三唑-1-基)苯甲酰氨基)癸基)三苯基氯化鏻(化合物9)
[0095]
将化合物8(120mg,0.322mmol)、化合物6(320mg,0.642mmol)、edc(123mg,0.642mmol)和et3n(0.22ml,1.6mmol)溶解于dmf(5ml)中,并将反应物搅拌2小时。粗产物通过滴加反应混合物至冰冷et2o(150ml)中来沉淀。将粗产物溶解于meoh(3ml)和柠檬酸(5%,3ml)和半饱和水性nh4cl(10ml)中,并用dcm(3x30ml)萃取。通过柱色谱(甲苯/甲醇,梯度100:5-100:20)获得式9的纯产物(34mg,13%),呈浅黄色泡沫状。
[0096]
[0097]1h nmr(600mhz,氯仿-d)δ10.55(s,1h),8.14(dd,j=7.8,1.7hz,1h),7.80(ddd,j=19.6,14.1,8.1hz,9h),7.69(td,j=7.7,3.3hz,6h),7.53
–
7.48(m,1h),7.46
–
7.29(m,7h),7.24(d,j=8.4hz,1h),7.19
–
7.11(m,1h),7.09(dd,j=7.9,1.1hz,1h),7.01(dd,j=8.2,1.1hz,1h),6.94(ddd,j=8.1,7.3,1.1hz,1h),6.81(t,j=7.5hz,1h),3.60(m,2h),3.19(q,j=6.6hz,2h),1.90
–
1.61(m,2h),1.60
–
1.48(m,2h),1.46
–
1.06(m,12h)。
[0098]
13
c nmr(151mhz,氯仿-d)δ166.54,160.31,156.76,156.47,153.04,135.03(d,j=2.9hz),134.96,134.22,133.51(d,j=9.9hz),132.01,131.06,130.43(d,j=12.5hz),128.96,128.56,127.36,126.26,119.43,119.11,118.30(d,j=85.8hz),117.69,116.94,113.88,112.87,39.77,29.69,29.52(d,j=16.0hz),28.33,28.04,28.03,27.92,27.66,25.93,22.15(d,j=33.7hz)。
[0099]
c49h50o3n4p
+
hrms计算值:773.36150,实测值:773.36134。
[0100]
实施例10
[0101]
(10-(2,4-双(2-羟苯基)-1h-咪唑-1-基)癸基)三苯基氯化鏻(化合物10)
[0102]
向2-溴-2
′‑
羟基苯乙酮(108mg,0.5mmol)在dmf(2ml)中的溶液一次性加入化合物6(138mg;0.303mmol))。将混合物加热至90℃持续3小时。然后加入水杨醛(122mg;1.0mmol)和乙酸铵(77mg;1.0mol)。将混合物加热过夜至(90℃)。tlc分析(chcl3/meoh/ch3cooh 100:10:1)指示形成复杂反应混合物,其中产物可以通过5%fecl3溶液可视化。然后将反应混合物冷却至室温并用二乙醚(20ml)稀释。在反应容器壁上形成棕色沉淀。倾析出溶剂并将沉淀溶解在甲醇(3ml)中。在将半饱和氯化铵溶液(15ml)加入粗产物的甲醇溶液中后,形成乳状乳液。将乳液通过二氯甲烷(3x 50ml)萃取,并将合并的有机层经mgso4干燥。然后将有机层在真空下浓缩,并经受柱色谱(10ml二氧化硅,甲苯/甲醇100:5-100:20)。式10的产物的合并级分以黄色油状物的形式得到(23mg,11%)。
[0103][0104]1h nmr(600mhz,氯仿-d)δ11.67(s,1h),8.12(dd,j=7.7,1.6hz,1h),7.83
–
7.70(m,10h),7.55(td,j=8.2,3.6hz,6h),7.54
–
7.39(m,2h),7.30(ddd,j=8.4,7.3,1.8hz,1h),7.30
–
7.20(m,2h),7.20
–
7.16(m,1h),6.83(ddd,j=7.1,6.4,≈1.0hz,2h),6.85(td,j=7.4,≈1.1hz,1h),4.15(t,j=7.4hz,2h),3.60(td,j=11.9,5.5hz,2h),1.99(p,j=7.5hz,2h),1.65
–
1.45(m,4h),1.38
–
1.27(m,2h),1.25
–
1.10(m,8h)。
[0105]
13
c nmr(151mhz,氯仿-d)δ157.20,154.88,153.75,142.61,135.72(d,j=3.1hz),132.33(d,j=11.5hz),133.54,131.62,129.12(d,j=12.5hz),127.47,126.8,119.48,117.40,117.16(d,j=87.1hz),116.90,116.22,114.28,113.66,48.49,30.69(d,j=16.3hz),重叠(28.12,28.11,28.05),26.00,23.00,22.38,22.28(重叠),21.12。
[0106]
实施例11
[0107]
(10-(4-(3,5-双(2-羟苯基)-1h-1,2,4-三唑-1-基)苯甲酰氨基)癸基)三苯基鏻
(化合物1)
[0108]
将化合物6(50mg,0.073mmol)溶解在无水dcm(1ml)中,并滴加tfa(1ml)。将反应混合物搅拌2.5小时,然后蒸发溶剂并将残余物溶解在乙醇(2ml)中。缓慢加入2-(2-羟苯基)-4h-苯并[e][1,3]噁嗪-4-酮(35mg,0.146mmol)至反应混合物,并滴加et3n(0.02ml,0.15mmol)。将反应混合物加热至70℃并搅拌2小时。蒸发溶剂。将混合物溶解于meoh中,并用饱和水性nh4cl和dcm萃取3次。通过硅胶柱色谱(12ml)(甲苯/甲醇,梯度0-10%甲醇)获得纯产物,呈浅黄色泡沫状。
[0109][0110]
实施例12
[0111]
测试了化合物1和比较性化合物地拉罗司(dfx)杀死/抑制代表雌激素依赖性乳腺癌类型的恶性乳腺癌mcf7细胞增殖的功效。简而言之,在一天将每孔10000个细胞接种在96孔板中,并在第二天加入测试化合物并与细胞一起孵育48小时。然后将细胞用4%多聚甲醛固定,用0.05%结晶紫染色,用pbs洗涤,并溶解在1%sds中。然后测量板在595nm处的吸光度,量化活细胞的数量。结果表明,化合物1与地拉罗司相比非常有效,并显示出细胞抑制作用,其与10μm地拉罗司相比,在低至8nm的浓度下将增殖抑制至50%(ic
50
),因此在低四个数量级的浓度下显示出相同的作用。比较示于表1中。
[0112]
表1:dfx和化合物1在孵育48小时后对恶性mcf7细胞中细胞存活率的作用,如通过结合细胞抑制作用以及细胞毒性作用的结晶紫测定所测量的。数据代表与未处理的对照相比的活细胞的平均百分比,而括号中的数字代表标准偏差。
[0113]
浓度化合物1dfx0μm100.00(8.82)100.00(8.82)0.030μm33.73(8.88)-0.100μm23.65(7.48)-0.300μm14.75(3.80)-1μm7.28(1.15)103.83(11.68)3μm5.20(2.53)76.03(12.85)10μm2.31(2.67)56.52(1.43)30μm0.69(0.23)23.89(7.38)ic
50
(μm)0.008(0.003)10.90(1.48)
[0114]
实施例13
[0115]
测试化合物1和比较性化合物地拉罗司杀死/抑制代表难以治疗的三阴性乳腺癌的恶性mda-mb-231癌细胞增殖的功效。程序与实施例12中的程序相同。结果表明,化合物1与地拉罗司相比非常有效,并且表明ic
50
值为131nm,相比之下地拉罗司为70μm,因此显示出
几乎三个数量级的差异。比较示于表2中。
[0116]
表2:dfx和化合物1在孵育48小时后对恶性mda-mb-231细胞中的细胞存活率的作用,如通过结合细胞抑制作用以及细胞毒性作用的结晶紫测定所测量的。数据代表与未处理的对照相比的活细胞的平均百分比,而括号中的数字代表标准偏差。
[0117]
浓度化合物1dfx0μm100.00(5.82)100.00(6.60)0.030μm71.10(7.60)-0.100μm52.47(8.87)-0.300μm37.42(5.70)-1μm27.81(2.09)109.15(14.65)3μm18.23(5.89)90.81(12.17)10μm0.29(1.46)70.38(2.35)30μm0.51(0.38)62.07(11.57)100μm-55.70(15.68)300μm-14.09(4.57)ic
50
(μm)0.131(0.019)70.02(15.26)
[0118]
实施例14
[0119]
测试了化合物1、2和对比化合物地拉罗司抑制增殖/杀死若干种乳腺、胰腺和卵巢来源的恶性细胞系(mcf7、mda-mb-231、bxpc3和ovcar3)的功效。程序与实施例12中的程序相同。结果表明选择性显著提高,如表3所记录。
[0120]
表3:dfx和化合物1和2在孵育48小时后对非恶性成纤维细胞(hfp1)和若干种恶性细胞系(mcf7、mda-mb-231、bxpc3和ovcar3)中的细胞存活力的作用,如通过结合细胞抑制作用以及细胞毒性作用的结晶紫测定所测量的。数据代表与未处理的对照相比的活细胞的平均百分比,而括号中的数字代表标准偏差。
[0121][0122]
化合物1显示了抑制所有测试癌细胞生长的能力,如表3所记录,描述了如在实施例12(测量联合的抗增殖和细胞死亡诱导的作用)中进行的用dfx或化合物1或2处理的细胞的结晶紫染色。重要的是,新合成的化合物1的体外ic
50
值与亲本dfx相比几乎低三个数量级,并且比其他线粒体靶向螯合剂低一至两个数量级,表明它是一种高活性化合物(表1、2),达到的功效与基于将如在其他线粒体靶向的衍生物(例如mitotax,wo2014/173374)中所见的母体药物的作用增强两个数量级的dfx的线粒体靶向所预期的功效相比更高。此外,化合物2显示了ic
50
值在100nm范围内,高于化合物1,但仍然非常强效。
[0123]
实施例15
[0124]
化合物1在10-100nm范围内的对癌细胞系显示出非常高的功效,如前表所示。这种作用似乎主要基于化合物1的细胞抑制作用。为了定义杀死细胞的能力,dfx和化合物1的细胞死亡诱导效力(细胞毒性功效)已经经由膜联蛋白v/pi染色进行测试,其通常用作细胞凋亡/坏死细胞死亡的量度。简而言之,将细胞以每孔100000个接种在12孔板中,与选定的化合物一起孵育48小时,然后旋转漂浮细胞,将贴壁细胞胰蛋白酶消化并也旋转,然后用膜联蛋白v-fitc和pi探针染色。然后用pbs洗涤细胞并经由荧光激活分选仪进行测量。表现出膜联蛋白v和/或pi阳性的细胞被认为是死细胞。如表4所示,在所有恶性乳腺癌细胞中,与dfx相比,化合物1显示出显著增强的诱导细胞死亡的能力。重要的是,非恶性hfp1细胞对化合物1的作用明显较不敏感,并且只有在高于5μm的浓度下才能检测到毒性作用,其中所有测试的癌细胞都表现出大量死细胞(表4)。
[0125]
测试本发明的化合物的生物作用并与已知的化合物-地拉罗司进行比较。本发明的最有活性的化合物杀死癌细胞,其作用比地拉罗司高出近3个数量级。这是前所未有的且非常出乎意料的。化合物2显示出ic
50
值在100nm的范围内,细胞毒活性显著更高,表明类似化学基团的其他化合物显示出类似的作用,并且本发明不仅限于地拉罗司衍生物。
[0126]
重要的发现是,本发明的化合物确实对非恶性细胞显示出较小的毒性作用并且在体内良好耐受,因此,它们在杀死癌细胞方面更具选择性。
[0127]
表4:48小时后如用膜联蛋白v/pi测定法所测量的化合物1、化合物2和dfx对恶性细胞(mcf7、bxpc3、ovcar、mda-mb-231)和非恶性细胞(hfp1、mrc5)中细胞死亡诱导的作用。数据代表死细胞的平均百分比,而括号中的数字代表标准偏差。
[0128][0129]
实施例16
[0130]
还经由用细胞分析仪juli fl进行实时监测,通过另一种方法测试了化合物1抑制癌细胞增殖的能力。简而言之,将mcf7细胞在前一天接种在6孔板中,以在第二天达到大约5%的汇合度。第二天,仪器的相机聚焦在具有大约5%汇合度的特定区域,并连续监测细胞,每30分钟记录汇合度和视觉外观,持续48小时。然后将值表示为实验结束时的简单汇合度,或表示为在给定时间段内代表汇合度的线的斜率(表5)。从收集到的数据中清楚地看出,在30nm浓度的化合物1下,确实看到了对细胞增殖的显著抑制。
[0131]
表5化合物1对恶性mcf7细胞中的细胞增殖的作用,通过经由juli fl连续监测细胞汇合度进行测量。数据代表细胞汇合度的平均值或测量相对于时间的相对细胞数的线的斜率的平均值,而括号中的数字代表两种情况下的标准偏差。
[0132][0133]
实施例17
[0134]
化合物1的施加导致细胞呼吸的显著抑制,如通过使用seahorse仪器的ocr测量所证明(表6)。对于seahorse分析,将20000个细胞接种在96孔板中并在测量前立即添加(添加,表6)或与化合物1一起孵育1小时(预孵育,表7)。之后,使用seahorse xfe96分析仪(agilent)在3分钟混合之前的三个3分钟循环中测量耗氧率(ocr)。结果表示三个循环的平均值。数据表明,这两种情况都导致ocr的显著抑制。
[0135]
表6立即添加化合物1对mcf7细胞中的耗氧率(ocr)的作用,如通过seahorse仪器所测量的。数字代表平均耗氧率(ocr;pmol*min-1 10000个细胞-1
),而括号中的数字代表标准偏差。
[0136]
添加对照5μm10μm基础71.5(23.9)52.4(9.2)49.6(9.6)质子泄漏20.1(11.1)43.5(7.6)37.3(6.1)最大80.4(26.4)29.7(5.8)26.3(5.4)
[0137]
表7如由seahorse仪器测量的在与化合物1一起预孵育1小时后,化合物1对mcf7细胞中的耗氧率(ocr)的作用。数字代表平均耗氧率(ocr;pmol*min-1 10000个细胞-1
),而括号中的数字代表标准偏差。
[0138]
1h预孵育对照5μm10μm基础77.1(23.8)39.9(5.8)30.9(3.1)质子泄漏19.2(10.2)33.1(4.7)25.9(3.1)最大78.9(22.2)20.7(4.5)17.4(3.8)
[0139]
实施例18
[0140]
使用特定的线粒体靶向探针mitosox(thermofisher scientific)在mcf7和mda-mb-231细胞中评价了化合物1对线粒体活性氧类(ros)产生的作用,该探针主要测量线粒体超氧化物并在结合后变为荧光。在所述时间间隔内用化合物1处理的细胞用探针染色,然后经由流式细胞术进行分析。数据显示化合物1能够诱导线粒体ros。
[0141]
表8 mitosox探针测量值代表平均荧光值,而括号中的数字代表标准偏差。
[0142][0143]
实施例19
[0144]
由于化合物1是一种铁螯合剂,我们已经评价了将化合物1与铁以2:1的比率预加载然后将其施加至细胞的效果,然后如实施例4一样使用膜联蛋白v/pi染色测量细胞死亡。此类预处理明显降低了由这种螯合剂诱导的细胞死亡的程度,证实铁螯合特性部分导致其在诱导细胞死亡中的作用,但可能其他一种或多种机制也参与了死亡诱导作用(表9)。
[0145]
表9:在孵育48小时后铁预加载对化合物1杀死癌细胞(mcf7、mda-mb-231)功效的作用,如与实施例4中一样进行的通过膜联蛋白v/pi染色所测量。数字代表死细胞的平均百分比,而括号中的数字代表标准偏差。
[0146][0147]
实施例20
[0148]
为了测试化合物1是否对抗性癌细胞有活性,使用了他莫昔芬抗性mcf7,t47d。用如实施例12中进行的结晶紫测定再次测量ic
50
值。已经证明,化合物1在对他莫昔芬敏感的亲代细胞和即使在他莫昔芬存在的情况下也能生长的抗性细胞之间诱导细胞抑制和细胞毒性作用的能力存在显著差异(表11),但ic
50
值仍在nm范围内,使这种化合物非常有效。该观察结果以及表明它在三阴性乳腺癌细胞系(诸如mba-mb-231)中也有效的数据表明,化合物1即使在难以治疗的癌症亚型中也具有活性。
[0149]
表10:化合物1对恶性(mcf7、t47d)细胞(亲本)及其在5μm他莫昔芬存在下生长的他莫昔芬抗性对应物(tam5r)中的细胞死亡诱导的作用。数据代表通过结晶紫测定获得的平均ic
50
值,而括号中的数字代表标准偏差。
[0150][0151]
实施例21
[0152]
为了测试化合物1对其他抗性癌细胞类型的存活力和增殖的作用,使用了可以在体外培养的癌症干细胞样细胞模型,即所谓的“乳腺球(mammospheres)”。这些细胞以3d微肿瘤的形式生长,并对许多已知的化疗药物表现出抗性。在该测定中,mcf7球被解离为单细胞/小球,在不粘附在mammocult培养基(stem cell technologies)中的特殊细胞培养塑料上每孔接种10000个细胞,并添加了1μm的化合物1和化合物2。这代表非细胞毒性浓度。使细胞生长并产生球140小时。然后通过cell titerglo 3d试剂(promega)测量细胞的量,该试剂确定3d培养细胞的细胞计数作为其atp含量的函数,该含量由化学发光测定法确定。可以看出,这两种化合物均能有效抑制肿瘤球的形成和生长,表明这些化合物对癌症干细胞样细胞具有活性。
[0153]
表11:化合物1和2对代表癌症干细胞样细胞模型的mcf7乳腺球的形成和生长的作用。在存在或不存在1μm的化合物1和2的情况下,使乳腺球生长140小时。形成的乳腺球的量化基于cell titer glo 3d测定法,该测定法经由发光测量细胞atp。数据代表标准化至设置为100的对照样品的平均化学发光atp值,而括号中的数字代表标准偏差。
[0154] 化合物1化合物20μm100(29.68)100(29.68)1μm6.61(1.91)2.82(0.28)
[0155]
实施例22
[0156]
为了限定细胞水平的作用,研究了化合物1和2在非细胞毒性药物浓度(1μm)下对细胞周期的作用。将细胞与化合物一起培养24和48小时,然后用与dna结合的vybrant dyecycle violetready flow试剂(thermofisher scientific)染色,并通过流式细胞术和kaluza程序进行分析以限定细胞周期的g1、s和g2/m期的比例。很明显,如果细胞周期,则这两种化合物都导致细胞在g1期中积累,其在以后的时间点变得更加突出(参见表12、13)。这表明所述化合物是细胞周期的强效抑制剂,解释了其细胞抑制活性的分子机制。
[0157]
表12化合物1和2对细胞周期分布的作用。将细胞在1μm化合物存在下培养24小时,然后用vybrant dyecycle violetready flow试剂染色并通过流式细胞术进行分析。使用kaluza软件进行细胞在每个细胞周期期中的比例。数据代表平均值,括号中的数字代表标准偏差。
[0158][0159]
表13化合物1和2对细胞周期分布的作用。将细胞在1μm化合物存在下培养48小时,然后用vybrant dyecycle violetready flow试剂染色并通过流式细胞术进行分析。使用kaluza软件进行细胞在每个细胞周期期中的比例。数据代表平均值,括号中的数字代表标准偏差。
[0160][0161]
实施例23
[0162]
在注射到nod/scidγ小鼠中的三阴性乳腺癌细胞(mda-mb-231)的异种移植模型中评估化合物1在体内对肿瘤生长的作用。每只小鼠皮下注射1x106个细胞。当肿瘤体积约为10mm3时,将动物随机分为两组,并每周两次腹膜内注射化合物1(在玉米油中2mg/kg)或媒介物(在玉米油中2.5%dmso)持续21天。使用vevo 3100preclinical imaging system(visualsonics)通过超声确定肿瘤大小。表14中显示的数据表明化合物1显著减慢了肿瘤生长。
[0163]
表14用媒介物或化合物1(2mg/kg,2次/周)处理的小鼠的肿瘤大小。值显示为与第0天(治疗开始)相比的平均相对肿瘤大小,括号中为标准偏差(n=5)。
[0164]
[0165]
实施例24
[0166]
在mcf7和mda-mb-231细胞中,使用测量几种活性类并在氧化后发出荧光的细胞渗透性ros探针2',7'-二氯二氢荧光素二乙酸酯(dcf-da;sigma-aldrich),评价化合物1对细胞活性氧类(ros)生成的作用。在所述时间间隔内用化合物1或化合物2(5μm)处理的细胞用探针染色,然后经由流式细胞术进行分析。表15中数据显示化合物1和化合物2显著提高细胞ros水平。
[0167]
表15 dcf-da探针测量值代表平均荧光值,而括号中的数字代表标准偏差。
[0168][0169]
实施例25
[0170]
使用报告蛋白keima在mcf7和mda-mb-231细胞中评估了化合物1和2对线粒体自噬诱导的作用。keima的激发光谱根据线粒体ph变化,在ph》6时在440nm处并在ph《5时在586处有一个激发峰,而第二激发峰在620nm处保持不变。因此,440nm/586nm荧光比的降低表明线粒体酸化。将用keima稳定转染的mcf7和mda-mb-231细胞与化合物1一起孵育24小时,并通过流式细胞术测量它们在两种激发波长下的荧光。表16中的数据表明化合物1诱导线粒体酸化,这是线粒体自噬诱导的明确标记。
[0171]
表16 keima 440nm/586nm荧光比的平均值,括号中为标准偏差。
[0172][0173]
实施例26
[0174]
测试化合物1和比较化合物地拉罗司(dfx)杀死/抑制若干种白血病细胞增殖的功效。简而言之,在一天将每孔10000个细胞接种在96孔板中,并在第二天加入测试化合物并与细胞一起孵育48小时。确定细胞存活率为降低探针alamarblue
tm
cell viability reagent的能力。结果表明,化合物1与地拉罗司相比非常有效,并显示出细胞抑制作用,导致在20-200nm浓度下增殖被抑制至50%(ic
50
),相比之下,对于地拉罗司为5-80μm,因此在
低40-4000倍的浓度下显示出相同的作用。比较显示于表17中。
[0175]
表17:化合物1和dfx在孵育48小时后对白血病细胞系的结合细胞抑制作用以及细胞毒性作用的存活力的作用,如用alamarblue
tm
cell viability reagent所测量的。数据代表与未处理的对照相比的活细胞的平均百分比,而括号中的数字代表标准偏差。
[0176][0177]
实施例27
[0178]
化合物1在20-200nm范围内对白血病细胞系显示出非常高的功效,如前表所示。这种作用似乎主要基于化合物1的细胞抑制作用。为了定义杀死细胞的能力,dfx和化合物1的细胞死亡诱导效力(细胞毒性功效)已经经由膜联蛋白v/pi染色进行了测试,该染色通常用作细胞凋亡/坏死细胞死亡的量度(参见实施例15)。如表18所示,在所有白血病癌细胞中,与dfx相比,化合物1表现出显著增强的诱导细胞死亡的能力。化合物1杀死癌细胞的作用比地拉罗司的作用高近3个数量级。这样的结果是前所未有的且非常出人意料的。
[0179]
表18 48小时后化合物1和dfx对白血病细胞系中细胞死亡诱导的作用,如用膜联蛋白v/pi测定法测量的。数据代表死细胞的平均百分比,而括号中的数字代表标准偏差。
[0180]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1