苯并嗪和邻苯二甲腈树脂的可固化组合物的制作方法
苯并嗪和邻苯二甲腈树脂的可固化组合物
1.相关申请的交叉引用
2.本技术要求2019年10月31日提交的美国临时专利申请号no.62/928,466的权益,其全部内容在这里作为参考明确引入。
3.关于联邦政府资助研究或开发的声明
4.不适用。
技术领域
5.本发明主要涉及包含邻苯二甲腈单体和含乙炔的苯并嗪化合物的可聚合热固性组合物以及它们在各种行业中的用途,例如但不限于建筑与结构、电子包装、能源和发电、航空航天、运输和医疗器械行业。
背景技术:
6.邻苯二甲腈单体是开发用于高温用途的一类新型高性能单体,如生产预浸料、层压板和结构复合部件。例如,us 6,420,464、us 8,039,576、us 8,853,343、us 9,920,165和美国专利公开no.2019/0047946公开了由酚类、芳族二醇与二苯基乙炔、来自可再生来源的多酚和双酚反应得到的各种邻苯二甲腈单体。已经发现,这些邻苯二甲腈单体固化后拥有优异的热稳定性和热氧化稳定性,初始分解温度大于450℃,并具有许多种其它极具吸引力的性能特点,如增强的阻燃性、热分解前不存在玻璃转化温度、高温下良好的机械性能、吸收率低、良好的耐腐蚀性和有利的uv屏蔽性能。
7.但由于单体前体的刚性和在最终固化产物中高的交联度,已知的是现有技术中邻苯二甲腈单体具有脆性。另外,这些邻苯二甲腈单体在室温下通常为固体,因此使用前必须熔化。再者,它们可能需要高于所希望的固化温度(例如大于250℃)和更长的时间来完全固化。
8.为了克服这些缺点,已经有尝试调节邻苯二甲腈单体单元之间部分的链长度,以降低它们的熔点并改进固化产物的柔韧性。还应用不同类型的催化剂来改进这些邻苯二甲腈单体的固化形为。最后,为了改进固化邻苯二甲腈产物的加工性能、固化行为或最终特性,us 5,939,508、wo 2017105890公开了与环氧树脂或苯并嗪树脂共聚的特定邻苯二甲腈单体,但这种组合物仍需要高于所希望的固化温度(例如远高于250℃)和更长的时间才能完全固化,并且对碳-碳复合材料的应用来说其残炭率不足。
9.希望进一步改进热固性组合物共聚中的固化循环,其中所述热固性组合物表现出更好的加工性和固化行为,并得到具有改进热和机械性能的固化产物。
技术实现要素:
10.本发明主要提供一种可聚合热固性组合物,其包含(i)含乙炔的苯并嗪化合物,和(ii)邻苯二甲腈单体。
11.本发明的可聚合热固性组合物可以固化形成具有改进的热和机械性能的热固性聚合物。因此,所述可聚合热固性组合物可以在各种应用中找到用途,例如但不限于建筑与结构、电子包装、军事、能源和发电、航空航天、运输和医疗器械行业。
具体实施方式
12.本发明主要提供一种可聚合热固性组合物,其包含(i)含乙炔的苯并嗪化合物,和(ii)邻苯二甲腈单体。已经令人惊奇地发现在可比条件下本发明的组合物比现有技术中其它的苯并嗪/邻苯二甲腈组合物固化更迅速。例如,本发明的组合物可能需要在温度至多200-220℃下分级固化,随后在温度250-260℃下后固化。作为对比,对于没有任何催化剂时的均聚来说,邻苯二甲腈单体本身通常需要至少350℃的固化温度(参见实施例3-8)。邻苯二甲腈单体能够与含乙炔的苯并嗪化合物中的乙炔官能团交联,导致改进的热和机械性能,例如增加的热稳定性、耐热性、残炭率和增强的结构刚性。不被任何特定理论所局限,据认为含乙炔的苯并嗪化合物的高焓值(例如大于800j/g)提供足够的热来促进邻苯二甲腈单体的聚合。同时,含乙炔的苯并嗪化合物在低于邻苯二甲腈单体熔点约20-40℃下也有能力溶解一定量的邻苯二甲腈单体,形成可易于通过尤其是输液应用处理的低粘度混合物。
13.如下术语应具有如下含义:
14.术语“包括”和其衍生形式不排除任何附加组分、步骤或过程的存在,不管它们是否在这里公开。为了避免任何疑义,如果没有相反指出,在这里应用术语“包括”要求的所有组合物均可以包括任何附加的添加剂或化合物。与之相比,如果在这里出现术语“主要由
…
组成”,除了那些对操作性不重要的以外,排除了任何后继列出的以外的任何其它组分、步骤或过程;和如果应用术语“由
…
组成”,则排除了没有具体描述或列出的任意组分、步骤或过程。如果不另外指出,术语“或”指所列的各元素及其任意组合。
15.在这里应用冠词“a”和“an”指冠词的语法宾语的一个或多个(即至少一个)。举例来说,“官能化的邻苯二甲腈单体”指一个官能化的邻苯二甲腈单体或多个官能化的邻苯二甲腈单体。
16.短语“在一个方面”、“按照一个方面”等通常指短语后的特定特征、结构或特性包括在本发明的至少一个方面中,和可以包括在本发明的多个方面中。重要的是,这种短语不必指同一方面。
17.如果说明书中称组分或特征“可以”、“会”、“可能”或“有可能”被包括或具有某一特性,则所述特定组分或特征不必被包括或具有所述特性。
18.按照一个方面,本发明提供一种可聚合热固性组合物,其包含含乙炔的苯并嗪化合物和邻苯二甲腈单体。
19.所述含乙炔的苯并嗪化合物在wo 1999/18092中有述,其内容在这里作为参考引入。具体地,含乙炔的苯并嗪化合物可以由单酚化合物、醛和伯胺的反应制备。
20.所述单酚化合物例如为但不限于苯酚、甲酚、2-溴-4-甲基苯酚、2-烯丙基苯酚、1,
4-氨基苯酚等。在一个实施方案中,所述酚类化合物为苯酚或烯丙基苯酚。
21.所述醛化合物可以为但不限于甲醛、低聚甲醛、聚甲醛或具有通式racho的化合物,其中ra为c
1-c
12
脂族基团。在一个实施方案中,所述醛化合物为甲醛。
22.所述伯胺可以为具有2-40个碳且具有一个或多个碳碳三键基团和任选的o、n、s或卤素杂原子的胺。伯胺的氮和碳碳三键基团之间的中间体任选地可以为c
1-c6烷基或具有6-12个碳的芳族基团,其中所述烷基任选被具有6-12个碳的芳族基团取代,所述芳族基团任选被c
1-c6烷基取代。所述碳碳三键基团包括具有如下通式的那些:-c≡crd、-ch
2-c≡crd、
[0023][0024]
其中rd为氢、任选被具有6-12个碳的芳族基团取代的c
1-c5烷基或任选被c
1-c5烷基取代的具有6-12个碳的芳族基团。在一个特定实施方案中,所述具有一个或多个碳碳三键的伯胺为3-氨基苯基乙炔和丙炔胺。
[0025]
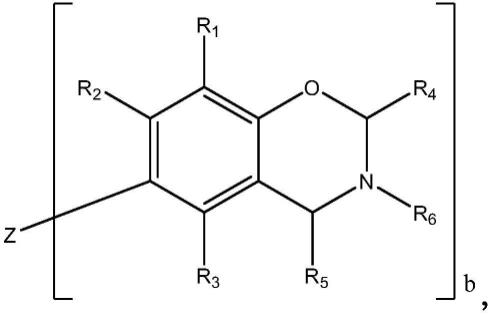
在一个实施方案中,所述一种或多种含乙炔的苯并嗪化合物按如下通式表示:
[0026][0027]
其中:r1、r2、r3、r4和r5各自独立地选自氢原子、取代或未取代的c
1-c
20
烷基、取代或未取代的c
2-c
20
链烯基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
2-c
20
杂芳基、取代或未取代的c
4-c
20
碳环基团、取代或未取代的c
2-c
20
杂环基团或c
3-c8环烷基;b为1-4的整数,其中:
[0028]
当b为1时,z为氢原子、取代或未取代的c
1-c
20
烷基、取代或未取代的c
2-c
20
链烯基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
2-c
20
杂芳基、取代或未取代的c
4-c
20
碳环基团、取代或未取代的c
2-c
20
杂环基团或c
3-c8环烷基;各r1为炔基取代的c
1-c
20
烷基、炔基取代的c
8-c
20
芳基、炔基取代的c
2-c
20
杂芳基、炔基取代的c
4-c
20
碳环基团、炔基取代的c
2-c
20
杂环基团或炔基取代的c
3-c8环烷基;
[0029]
当b为2时,z为直接键或取代或未取代的c
1-c
20
烷基、取代或未取代的带有芳基或杂芳基桥的c
2-c
20
烷基、取代或未取代的c
2-c
20
链烯基、取代或未取代的c
2-c
20
炔基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
2-c
20
杂芳基、o、s、s=o、o=s=o、c=o或c=ccl2;和
[0030]
当b为3或4时,z为取代或未取代的c
1-c
20
烷基、取代或未取代的带有芳基或杂芳基桥的c
2-c
20
烷基、取代或未取代的c
2-c
20
链烯基、取代或未取代的c
2-c
20
炔基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
2-c
20
杂芳基;和
[0031]
当b为3或4时,z为取代或未取代的c
1-c
20
烷基、取代或未取代的带有芳基或杂芳基
桥的c
2-c
20
烷基、取代或未取代的c
2-c
20
链烯基、取代或未取代的c
2-c
20
炔基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
2-c
20
杂芳基;和
[0032]
各r6为炔基取代的c
1-c
20
烷基、炔基取代的c
8-c
20
芳基、炔基取代的c
2-c
20
杂芳基、炔基取代的c
4-c
20
碳环基团、炔基取代的c
2-c
20
杂环基团或炔基取代的c
3-c8环烷基。
[0033]
在一个特定的实施方案中,含乙炔的苯并嗪化合物为苯酚3-氨基苯基乙炔苯并嗪或2-烯丙基苯酚3-氨基苯基乙炔苯并嗪。
[0034]
邻苯二甲腈单体由如下物质的反应获得:(i)多官能的酚类化合物和(ii)4-硝基邻苯二甲腈。
[0035]
所述多官能的酚类化合物可以为但不限于间苯二酚、双酚a、双酚c、双酚f、双酚e、双酚s、2,2
′
,6,6
′‑
四甲基双酚f、1,2,2,2-四酚基乙烷、硫代二苯酚、酚酞、二环戊二烯基二苯酚、1,8-羟基蒽醌、1,6-二羟基萘、2-2
′‑
二羟基偶氮苯、1,3,5-三羟基苯和包含至少一个呋喃基或噻吩基的多元酚化合物。
[0036]
在一个特定的实施方案中,所述多官能的酚类化合物选自双酚m、双酚a、双酚c、双酚p、2,2
′
,6,6
′‑
四甲基双酚f或呋喃基取代的2,2
′
,6,6
′‑
四甲基双酚f。
[0037]
所述包含至少一个呋喃基或噻吩基的多元酚化合物包括由酚类化合物和通式(1)的化合物获得的那些化合物:
[0038][0039]
其中x为氧或硫,q为氢或c
1-c5烷基,和j为1-3的整数。所述通式(1)的化合物包括但不限于糠醛、3-糠醛、3-甲基糠醛、5-甲基糠醛、5-乙基糠醛、2-噻吩-甲醛、3-噻吩-甲醛、3-甲基-2-噻吩-甲醛等。
[0040]
所述酚类化合物可以包括但不限于苯酚、甲酚、二甲苯酚(二甲基苯酚)如2,6-二甲苯酚、三甲基苯酚、2,5-烷基苯酚如2-叔丁基-5-甲基-苯酚或2-叔丁基-4-甲基苯酚、烯丙基苯酚、炔基苯酚、辛基苯酚、苯基苯酚、二苯基苯酚、愈创木酚、氢醌、间苯二酚、儿茶酚、萘酚、二羟基萘、甲基萘酚、双酚a、双酚f等。
[0041]
所述包含至少一个呋喃基或噻吩基的多元酚化合物可以通过本领域熟练技术人员通常已知的方法制备。例如,在碱和任选的醇或单取代的苯的存在下在温度约30-150℃或约60-90℃下酚类化合物可以与通式(1)的化合物缩合。通常,在缩合过程中存在的酚类化合物和通式(1)的化合物的量可以为每1摩尔通式(1)的化合物约1.5-20摩尔的酚类化合物。在一些实施方案中,缩合过程中存在的酚类化合物与通式(1)的化合物的量可以为每1摩尔通式(1)的化合物约1.8-10摩尔的酚类化合物。
[0042]
可以应用的碱的实例包括但不限于:碱金属氢氧化物,如氢氧化锂、氢氧化钠、氢氧化钾等;碱土金属氢氧化物,如氢氧化镁、氢氧化钙等;碱金属醇盐,如甲醇钠、乙醇钠、甲醇钾、乙醇钾、叔丁醇钾等;和碱土金属醇盐,如甲醇镁、乙醇镁等。这些碱可以单独使用或
两种或更多种组合使用。这些碱的用量可以为每1摩尔酚类化合物约0.005-2.0摩尔或每1摩尔酚类化合物约0.01-1.1摩尔。
[0043]
可以应用的醇或单取代的苯溶剂包括但不限于甲醇、乙醇、丙醇、异丙醇、甲苯、二甲苯等,它们可以单独使用或作为混合物使用。当需要时,这种溶剂的用量可以为每100重量份酚类化合物约5-500重量份或每100重量份酚类化合物约10-300重量份。
[0044]
可以通过向酚类化合物和通式(1)的化合物(和任选的醇或单取代的苯溶剂)的混合物中加入碱,并加热所得混合物来实施反应。替代地,可以在加热的条件下向酚类化合物和碱(和任选的醇或单取代的苯溶剂)的混合物中加入通式(1)的化合物。反应时间可以为约5-100小时。反应运行完成后,可以中和反应混合物。可以随后通过过滤或在真空下加热脱除任何的未反应物质。
[0045]
按照一个实施方案,所述包含至少一个呋喃基或噻吩基的多元酚化合物为选自通式(2)-(10)的化合物:
[0046][0047][0048]
其中n为约3-3.2的整数。
[0049]
在另一个实施方案中,包含至少一个呋喃基或噻吩基的多元酚化合物由双酚a或双酚f和通式(1)的化合物衍生得到,其中x为氧,和q和j按如上所定义。
[0050]
按照一个实施方案,通过在催化剂和任选溶剂的存在下使多官能的酚类化合物与4-硝基邻苯二甲腈反应,形成邻苯二甲腈单体。
[0051]
催化剂的实例包括但不限于上述的碱,以及碱金属盐如碳酸铯、碳酸钾或碳酸钠,有机锂试剂如甲基或正丁基锂,grignard试剂,或它们的任意组合。
[0052]
可以应用的溶剂的实例包括但不限于任何极性或非极性溶剂,如丙酮、乙腈、醇、
甲基乙基酮、甲基异丁基酮、二甲基甲酰胺、n-甲基吡咯烷酮、二甲基亚砜、六甲基磷酰胺或它们的组合。
[0053]
在另一个实施方案中,溶剂可以为能够与水形成共沸物的溶剂,如甲苯或二甲苯。已经令人惊奇地发现可以应用这些溶剂来帮助脱除在形成反应混合物的化合物(即包含至少一个呋喃或噻吩基的多元酚化合物、4-硝基邻苯二甲腈和碱)中存在的水,以及在包含至少一个呋喃基或噻吩基的多元酚化合物与4-硝基邻苯二甲腈的反应过程中形成的水。通过从溶剂和水的混合物中再结晶可以纯化邻苯二甲腈单体,以富集所得产物中的单体含量。
[0054]
按照另一个实施方案,所述包含至少一个呋喃基或噻吩基的多元酚化合物和所述官能化的邻苯二甲腈单体可以在相同的反应容器中形成,以提高整体过程时间和效率。在这种实施方案中,在第一步中,按如上所述在反应容器中形成包含至少一个呋喃基或噻吩基的多元酚。在第二步中,向反应容器中的包含至少一个呋喃基或噻吩基的多元酚中加入4-硝基邻苯二甲腈,以形成官能化的邻苯二甲腈单体。在第一步和第二步的反应中应用的碱、催化剂和溶剂可以相同或不同。在一些实施方案中,溶剂为甲苯或二甲苯。
[0055]
权利要求1的热固性组合物,其中邻苯二甲腈单体为具有如下通式的化合物:
[0056][0057]
其中r1至r
14
各自独立地选自氢原子、取代或未取代的c
1-c
20
烷基、取代或未取代的c
2-c
20
链烯基、取代或未取代的c
2-c
20
炔基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
2-c
20
杂芳基;和z选自直接键、取代或未取代的c
1-c
20
烷基、取代或未取代的带有芳基或杂芳基桥的c
2-c
20
烷基、取代或未取代的c
2-c
20
链烯基、取代或未取代的c
2-c
20
炔基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
2-c
20
杂芳基、o、s、s=o、o=s=o、c=o、c(=o)o或c=ccl2,或聚合物链的每个重复单元中含氧的聚合物链,包括但不限于聚醚、聚醚砜或聚醚酮。
[0058]
按照一个实施方案,所述可聚合热固性组合物按一种或多种含乙炔的苯并嗪化合物与邻苯二甲腈单体重量比为约1:1-10:1、或约1.5:1-10:1或约2:1-10:1包含一种或多种含乙炔的苯并嗪化合物和邻苯二甲腈单体。
[0059]
在可聚合热固性组合物中邻苯二甲腈单体的存在量基于可聚合热固性组合物总重量计可以为至少约1wt%、至少约5wt%、或至少约10wt%、或至少约20wt%、或至少约30wt%、或至少约40wt%、或至少约50wt%、或至少约60wt%、或至少约70wt%、或至少约80wt%、或至少约90wt%、或至少约99wt%。在其它实施方案中,在可聚合热固性组合物中邻苯二甲腈单体的存在量基于可聚合热固性组合物总重量计可以为约1-99wt%、或约5-90wt%、或约10-80wt%、或约20-70wt%、或约30-60wt%。
[0060]
本发明的可聚合热固性组合物可以通过将可聚合热固性组合物充分加热形成热固性聚合物固化。可以应用固化剂来加速热固性聚合物的热固性成型。因此,按照另一个实施方案,所述可聚合热固性组合物还包含固化剂。
[0061]
可以应用的固化剂包括但不限于芳族胺、伯胺、仲胺、二胺、多胺、胺取代的磷腈、
酚类、强酸、有机酸、强有机酸、无机酸、金属、金属盐、金属盐水合物、金属化合物、含卤素的芳族胺、粘土和化学改性的粘土。应用粘土或化学改性粘土可以改进热固性物质的机械和可燃特性。通常,粘土的化学改性包括用铵替换钠离子形成季铵盐。
[0062]
具体的固化剂包括但不限于双(4-(4-氨基苯氧基)苯基砜(p-baps)、双(4-(3-氨基苯氧基)苯基砜(m-baps)、1,4-双(3-氨基苯氧基)苯(p-apb)、1,12-二氨基十二碳烷、二苯胺、环氧胺固化剂、1,6-己二胺、1,3-苯二胺、1,4-苯二胺、p-甲苯磺酸、碘化亚铜、溴化亚铜、1,3-双(3-氨基苯氧基)苯(m-apb)、3,3'-二甲基-4,4'-二氨基二苯基砜、3,3'-二乙氧基-4,4'-二氨基二苯基砜、3,3'-二羧基-4,4'-二氨基二苯基砜、3,3'-二羟基-4,4'-二氨基二苯基砜、3,3'-二磺基-4,4'-二氨基二苯基砜、3,3'-二氨基二苯甲酮、4,4'-二氨基二苯甲酮、3,3'-二甲基-4,4'-二氨基二苯甲酮、3,3'-二甲氧基-4,4'-二氨基二苯甲酮、3,3'-二羧基-4,4'-二氨基二苯甲酮、3,3'-二羟基-4,4'-二氨基二苯甲酮、3,3'-二磺基-4,4'-二氨基二苯甲酮、4,4'-二氨基二苯基乙基氧化膦、4,4'-二氨基二苯基苯基氧化膦、双(3-氨基苯氧基-4'-苯基)苯基氧化膦、亚甲基二苯胺、六(4-氨基苯氧基)环三磷腈、3,3'-二氯-4,4'-二氨基二苯基砜、2,2'-双(三氟甲基)-4,4'-二氨基二苯基、2,2'-双(4-氨基苯基)六氟丙烷、双[4-(4-氨基苯氧基)苯基]2,2
′‑
六氟丙烷、1,1-双(4-氨基苯基)-1-苯基-2,2,2-三氟乙烷、3,3'-二氯-4,4'-二氨基二苯甲酮、3,3'-二溴-4,4'-二氨基二苯甲酮、苯胺-2-磺酸、8-苯胺-1-萘磺酸、苯磺酸、丁基磺酸、10-樟脑磺酸、2,5-二氨基苯磺酸、6-二甲基氨基-4-羟基-2-萘磺酸、5-二甲基氨基-1-萘磺酸、4-羟基-3-亚硝基-1-萘磺酸四水合物、8-羟基喹啉-5-磺酸、甲基磺酸、苯硼酸、1-萘磺酸、2-萘磺酸、1,5-萘二磺酸、2,6-萘二磺酸、2,7-萘二磺酸、苦基磺酸水合物、2-吡啶乙烷磺酸、4-吡啶乙烷磺酸、3-吡啶磺酸、2-吡啶基羟基甲烷磺酸、氨基苯磺酸、2-磺基苯甲酸水合物、5-磺基水杨酸水合物、2,4-二甲基磺酸、含磺酸的染料、有机含磷酸、苯基次膦酸、二苯基次膦酸、丙基膦酸、1-氨基乙基膦酸、4-氨基苯基膦酸、丁基膦酸、叔丁基膦酸、2-羧基乙基膦酸、2-氯乙基膦酸、二甲基膦酸、乙基膦酸、亚甲基二膦酸、甲基膦酸、磷酰基乙酸、双(羟基甲基)膦酸、氯甲基膦酸、二-正丁基膦酸、二氯甲基膦酸、二苯基二硫代膦酸、1,2-亚乙基二膦酸、n-hystaderyl膦酸、羟基甲基膦酸、正十八烷基膦酸、正辛基膦酸、苯基膦酸、亚丙基二膦酸、正十四烷基膦酸、浓硫酸、苯基膦酸、铜、铁、锌、镍、铬、钼、钒、铍、银、汞、锡、铅、锑、钙、钡、锰、镁、钴、钯、铂、溴化亚铜、氰化亚铜、铁氰化亚铜、氯化锌、溴化锌、碘化锌、氰化锌、亚铁氰化锌、乙酸锌、硫化锌、氯化银、氯化亚铁、氯化铁、铁氰化亚铁、氯化亚铁铂酸盐、氯化亚铁、硫酸亚铁、氯化钴、硫酸钴、氰化钴、氯化镍、氰化镍、硫酸镍、碳酸镍、氯化锡、氯化亚锡水合物、氯化亚锡二水合物、硝酸铝水合物、硝酸铝九水合物、三苯基氧化膦复合物、蒙脱石、化学改性的蒙脱石、4,4
′‑
(1,3-亚苯基二氧)二苯胺、4,4
′‑
(1,4-亚苯基二氧)二苯胺、双(4-(4-氨基苯氧基)苯基]砜、4,4
′‑
(4,4
′‑
亚异丙基二苯基-1,1
′‑
二基二氧)二苯胺、4,4
′‑
(1,3-亚苯基二亚异丙基)二苯胺、4,4
′‑
(1,4-亚苯基二亚异丙基)二苯胺、4,4
′‑
(1,1
′‑
联苯基-4,4
′‑
二基二氧)二苯胺、4,4
′‑
亚甲基二苯胺、4,4
′‑
磺酰基二苯胺、4,4
′‑
亚甲基-双(2-甲基苯胺)、3,3
′‑
亚甲基二苯胺、3,4
′‑
亚甲基二苯胺、4,4
′‑
氧二苯胺、4,4
′‑
(亚异丙基)二苯胺、4,4
′‑
(六氟亚异丙基)二苯胺、4,4
′‑
(六氟亚异丙基)双(p-亚苯基氧)二苯胺、4,4
′‑
二氨基二苯甲酮、如下化合物:
[0063][0064]
以及它们的混合物。
[0065]
固化剂在可聚合热固性组合物中的存在量可以为可聚合热固性组合物总重量的至少约0.5wt%、或至少约1wt%、或至少约2wt%、或至少约5wt%、或至少约10wt%、至少约15wt%或甚至至少约20wt%。
[0066]
在其它实施方案中,固化剂的存在量可以为可聚合热固性组合物总重量的小于约40wt%、或小于约35wt%、或小于约30wt%、或小于约25wt%。在其它实施方案中,固化剂的存在量可以为可聚合热固性组合物总重量的约0.25-45wt%或约1-40wt%。
[0067]
在低于熔点的温度(例如但不限于比组分的熔点低35-40℃或更多的温度)下,通过应用常规设备如搅拌容器、搅拌棒、球磨机、样品混合器、静态混合器或带式混合器混合含乙炔的苯并嗪化合物和邻苯二甲腈单体以形成热固性组合物,可以制备可聚合热固性组合物。
[0068]
然后可以固化可聚合热固性组合物,以形成热固性聚合物。正如这里所应用,短语“固化”指上述热固性组合物转化为不溶的和不熔的交联产物,同时成形以得到如模板、拉挤板或层压板等的成形制品,或者得到如涂层、搪瓷或粘结的两维结构。典型的固化方法包括环境温度固化至应用热、辐射或各种能源的任意组合的高温固化。典型的固化温度可以为约50-500℃,如约75-375℃或约80-300℃,固化足够时间以至少部分或基本上或完全固化所述组合物,例如3-20小时,或约5-15小时,或约6-15小时,或约7-12小时,或约8-10小时。
[0069]
另外,可以在一个或多个固化步骤中实施固化。例如,可以使所述可聚合热固性组合物在温度约50-500℃(如约75-375℃或约80-300℃)下经受初始固化足够时间以至少部分固化所述组合物,例如3-10小时的时间,然后在温度低于300℃或在280℃或低于280℃和甚至在260℃或低于260℃后固化足够时间,以基本上或完全固化所述组合物,例如2-4小时的时间。固化前,所述可聚合热固性组合物在75℃下的粘度小于3000厘泊、或小于1000厘泊、或小于500厘泊、或小于175厘泊。
[0070]
按照一个实施方案,本发明的可聚合热固性组合物还可以包括附加的单官能的苯并嗪或多官能的苯并嗪或它们的组合以及任意一种或多种固化剂和任选的添加剂或第二种邻苯二甲腈单体以形成另一种热固性组合物。所述添加剂可以包括但不限于:填料c14
杂环基、c
6-c
14
芳基、c
6-c
14
杂芳基、卤素、氰基、硝基、硝酮、氨基、酰胺基、酰基、氧酰基、羧基、氨基甲酸根、磺酰基、磺酰胺和硫酰基。
[0080]
按照一个实施方案,上述多官能的苯并嗪化合物为由多官能的酚类化合物、醛如甲醛和伯胺的反应获得的化合物。
[0081]
所述多官能的酚类化合物可以为但不限于间苯二酚、双酚a、双酚f、双酚e、双酚s、1,2,2,2-四苯酚乙烷、硫代二苯酚、酚酞、二环戊二烯基二苯酚、1,8-羟基蒽醌、1,6-二羟基萘、2-2
′‑
二羟基偶氮苯和1,3,5-三羟基苯。在一个特定的实施方案中,所述多官能的酚类化合物为4
′
4-二苯酚或4,4-硫代二苯酚。
[0082]
所述伯胺可以为但不限于如上所述的具有至少一个碳碳三键基团的那些伯胺,以及苯胺、o-、m-和p-亚苯基二胺、联苯胺、4,4
′‑
二氨基二苯基甲烷、环己胺、1,4-二氨基环癸基(cyclohydexyl)、丁基胺、甲基胺、己基胺、烯丙基胺、糠基胺、亚乙基二胺、亚丙基二胺和二氨基二苯基砜。在一个特定的实施方案中,所述伯胺为苯胺和糠基胺。
[0083]
在另外一个实施方案中,提供一种热固性聚合物,其通过使任何合适的基质与如上所述任意一种可聚合热固性组合物接触、并使所述基质/可聚合热固性组合物经受热、辐射或各种能源的组合来固化所述基质/可聚合热固性组合物而获得。在一个实施方案中,可以应用本发明的可聚合热固性组合物将一种或多种基质粘结在一起,这通过使待粘接的相同或不同基质的一个或多个表面与可聚合热固性组合物在足以固化所述可聚合热固性组合物的条件下接触来实现。
[0084]
在一个替代的实施方案中,通过固化本发明的可聚合热固性组合物,可以由工业上公知的技术如拉挤、灌注、模塑、包封或涂覆获得复合制品。因此,本发明的可聚合热固性组合物可用于生产复合制品的方法中,如铸件、预浸料、粘合板、层压板和金属箔包覆的层压板。
[0085]
对于某些用途来说,通过加入增强纤维可以调节所述复合制品的特性。增强纤维的实例包括玻璃、石英、碳、氧化铝、陶瓷、金属、芳纶、天然纤维(如亚麻、黄麻、剑麻、大麻)、纸、丙烯酸和聚乙烯纤维和它们的混合物。增强纤维可以为各种模式中的任意一种,例如通过在一个方向上并联连续纤维或不连续纤维(短纤维)形成的束或粗纱、布如编织物或编织垫、辫状物、单向、双向、随机、拟各向同性或三维分散的垫类材料、非均质格子或网状材料,和三维材料如三轴机织物。
[0086]
因此,在另一个实施方案中,提供一种生产复合制品的方法,包括如下步骤:使增强纤维层与可聚合热固性组合物接触,以涂覆和/或浸渍所述增强纤维;和固化所述涂覆和/或浸渍的增强纤维以得到复合制品。
[0087]
涂覆和/或浸渍可以通过湿式或热熔方法实施。在湿式方法中,首先将热固性组合物溶解于溶剂中以降低粘度,随后实施增强纤维的涂覆和/或浸渍,并应用烘箱等蒸发掉溶剂。
[0088]
在热熔方法中,可以通过用可聚合热固性组合物直接涂覆和/或浸渍增强纤维来实施涂覆和/或浸渍,可以将所述热固性组合物加热以降低其粘度,或者替代地,首先在剥离纸等上得到可聚合热固性组合物的涂覆膜,和将所述膜放置在增强纤维的一面或两面上,并施用热和压力以实施涂覆和/或浸渍。
[0089]
按照另一个方面,提供一种在rtm系统中生产复合制品的方法。所述方法包括如下
中):154.2,148.2,129.2,127.9,126.7,125.0,122.9,121.4,120.9,120.56,118.6,117.0,83.8,78.8,77.1,50.2.lcms:236.1074(m+h
+
,calc.236.1075)。
[0099]
向铝盘中加入14克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa)。然后将所述铝盘放入80℃真空烘箱中熔化和脱气1小时。脱气后,所述材料在如下条件下逐级固化:120℃2小时、150℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0100]
实施例2:2-烯丙基苯酚3-氨基苯基乙炔苯并嗪(烯丙基ph-apa)的合成和均聚
[0101][0102]
向装有温度计、冷凝器和氮气入口的1000ml的四颈圆底烧瓶中加入2-烯丙基苯酚(134g,1.0mol)、低聚甲醛(62.9g,2.1mol)和甲苯(205ml)和丁醇(34ml)。将混合物加热至70℃,和然后向搅拌的混合物中加入3-氨基苯基乙炔(117g,1.0mol)。然后将所得混合物加热至100℃,以通过共沸过程脱除大部分水。再将反应混合物加热至110℃以完成过程。过滤后,产物用3n氢氧化钠洗涤,然后用水洗涤,并干燥。在真空下脱除溶剂后,产物在真空中在100℃下再干燥3小时,作为棕色液体得到250g(90%)产物。通过gpc分析确定主要组分单体约76%、二聚物约12%、三聚物约5%。主要组分的1h nmr(ppm,cdcl3)为:7.10-7.30(m,4h),6.90-7.00(m,2h),6.81(dd,1h),5.88-5.96(m,1h),5.43(s,2h),4.95-5.05(m,2h),4.62(s,2h),4.08(s,1h),3.24(d,2h);
13
c nmr(ppm,cdcl3):151.7,147.9,136.5,129.6,128.1,127.9,127.1,125.3,123.8,122.4,120.7,120.1,120.0,118.1,115.6,83.8,80.1,78.2,49.4,31.3;lc-ms:276.1388(m+h
+
;calc.276.1388)。
[0103]
向铝盘中加入14克2-烯丙基苯酚3-氨基苯基乙炔苯并嗪(allph-apa)。然后将所述铝盘放入80℃真空烘箱中熔化和脱气1小时。脱气后,所述材料在如下条件下逐级固化:120℃2小时、150℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0104]
实施例3:2,2
′
,6
′6′‑
四甲基双酚f邻苯二甲腈(tmbf-pn)的合成和均聚
[0105][0106]
向装有温度计、冷凝器和氮气入口的1000ml的四颈圆底烧瓶中加入2,2
′
,6,6
′‑
四甲基双酚f(100g,0.39mol)、4-硝基邻苯二甲腈(135.1g,0.78mol)和dmf(350ml)。然后加入k2co3(148.6g,1.05mol)。将所得混合物加热至80-90℃持续2-3小时。将混合物冷却至环境温度,并倒入稀hcl中进行沉淀。沉淀物通过过滤收集,用水洗涤至中性,之后用甲醇洗涤。真空干燥固体,作为棕褐色固体得到194g(98%)粗产物,在254nm下通过hplc分析确定纯度
为95%。由乙腈进一步重结晶得到灰白色粉末状产物,纯度大于97%。在10℃/min下在191-193℃下dsc扫描确定熔点至多400℃。1h nmr(ppm,cdcl3):7.733(d,2h),7.14-7.17(m,4h),7.01(s,4h),3.91(s,2h),2.21(s,2h),1.99(s,12h);
13
c nmr(ppm,cdcl3):161.164(2c),147.872(2c),139.112(2c),135.652(2c),130.582(4c),130.010(4c),119.823(2c),119.660(2c),117.744(2c),115.498(2c),115.130(2c),108.246(2c),40.690(1c),16.192(4c);ftir(cm-1)3105.88,3077.27,3042.23,2949.33,2916.23,2860.75,2231.82,1592.23,1567.61,1479.62,1444.10,1420.06,1411.25,1382.71,1372.87,1329.94,1307.01,1277.14,1244.61,1191.46,1162.48,1134.01,1085.95,1020.57,980.57,972.99,961.75,949.15,904.55,895.84,885.14,868.92,848.79,840.06,808.47,765.01,757.67,727.54,720.58,688.45,662.06;lc-ms:c33h24n4o2(mw:508,98.16%)。
[0107]
向铝盘中加入14克邻苯二甲腈单体。然后将铝盘放入200℃烘箱中以熔化所述材料。随后,所述材料在如下条件下逐级固化:260℃6小时、300℃3小时和350℃4小时。也确定该固化产物的dma和tga。结果示于下表1和2中。
[0108]
实施例4:呋喃基-2,2
′‑
6,6
′‑
四甲基双酚f邻苯二甲腈(ftmbf-pn)的合成和均聚
[0109][0110]
步骤1:向配备有机械搅拌器和回流冷却器的500ml四颈圆底烧瓶中加入61.08克2,6-二甲苯酚和32.04克甲醇。然后加入2克氢氧化钠并搅拌溶解。加热回流所得混合物,并在回流下在2小时的时间间隔中逐滴加入24.0克糠醛。然后所述混合物再回流15小时,并通过hplc监测转化完成,随后用35克20%的磷酸二氢钠水溶液中和所述混合物。沉淀的晶体通过过滤收集,用1:1的甲醇/水溶液洗涤,并在真空干燥烘箱中干燥。得到72.6克四甲基双酚呋喃(90.8%)。hplc确定产物非常纯(99.7%)。
[0111][0112]
步骤2:向配备有温度计、带有冷凝器的dean-stark分水器和氮气入口的1000ml四颈圆底烧瓶中加入来自步骤1的呋喃基-四甲基双酚(32.2克,0.1摩尔)、粉末状的k2co3(33.2克,0.24摩尔)、甲苯(100ml)和n,n-二甲基甲酰胺(dmf)(146.1克)。用氮气使所得混合物脱气,并在140℃下加热所述混合物回流10-12小时。然后通过精馏脱除甲苯,并将反应混合物冷却至30℃。然后一次性加入4-硝基邻苯二甲腈(35.3克,0.204摩尔),并在80℃加热反应混合物,用hplc监测转化完成。然后将混合物冷却至环境温度,并倒入冷的去离子水中,从而形成固体。沉淀晶体通过过滤收集,用冷去离子水洗涤至中性,然后用1:1的甲醇/水溶液洗涤。得到的深黄色固体真空干燥,得到54.5克(95%)的标题产物。在10℃/min下通过dsc扫描确定产物熔点为219.6℃。1h nmr(ppm,cdcl3):7.733(d,2h),7.437(dd,1h),7.13-7.17(m,4h),7.00-7.03(s,4h),6.374(dd,1h),6.036(d,1h),5.39(s,1h),2.09(s,12h);
13
c nmr(ppm,cdcl3):160.981(2c),155.809(1c),148.253(2c),142.181(1c),
139.815(2c),135.694(2c),130.663(4c),129.794(4c),119.817(2c),119.714(2c),117.689(2c),115.499(2c),115.123(2c),110.266(1c),108.500(1c),108.280(2c),49.745(1c),16.296(4c);ftir(cm-1)3111.2,3075.5,3043.7,2963.5,2921.2,2862.4,2231.2,1673.8,1582.7,1564.0,1478.0,1421.8,1310.4,1276.7,1244.7,1182.0,1163.2,1133.4,1087.2.80,1011.5,950.0,880.4,835.6,781.3,734.8。
[0113]
向铝盘中加入14克邻苯二甲腈单体。然后将铝盘放入220℃烘箱中以熔化所述材料。随后,所述材料在如下条件下逐级固化:260℃6小时、300℃3小时和350℃4小时。也确定该固化产物的dma和tga。结果示于下表1和2中。
[0114]
实施例5:双酚a邻苯二甲腈(bisa-pn)的合成和均聚
[0115][0116]
向装有温度计、冷凝器和氮气入口的1000ml的四颈圆底烧瓶中加入双酚a(80g,0.35mol)、4-硝基邻苯二甲腈(121.3g,0.7mol)和dmf(300ml)。然后加入粉碎的k2co3(133.5g,1.05mol)。将所得混合物加热至80-90℃持续2-3小时。冷却混合物至环境温度,并倒入稀hcl中以形成固体。沉淀物通过过滤收集,用水洗涤至中性,之后用甲醇洗涤。真空干燥固体,作为绿色粉末得到166g(99%)粗产物,在280nm下通过hplc分析确定纯度约93%。由乙腈进一步重结晶得到绿色晶体产物(82.5%的收率),纯度超过97%。在10℃/min下通过dsc扫描确定熔点为198℃。1h nmr(ppm,cdcl3):7.733(d,2h),7.33-7.36(m,4h),7.27-7.29(m,4h),7.00-7.03(m,4h),3.91(s,2h),2.21(s,2h),1.759(s,6h);
13
c nmr(ppm,cdcl3):161.818(2c),151.584(2c),148.304(2c),135.419(2c),128.979(4c),121.619(2c),121.364(2c),120.223(4c),117.597(2c),115.401(2c),115.043(2c),108.796(2c),42.707(1c),30.938(2c);ftir(cm-1)3074.13,3050.53,2973.19,2935.48,1876.84,2233.15,1915.12,1669.92,1589.05,1560.79,1502.11,1485.78,1422.65,1407.54,1388.51,1366.70,1305.39,1288.35,1251.36,1209.84,1176.79,1163.28,1112.99,1103.00,1081.80,1016.02,952.72,922.78,903.99,887.50,854.97,854.07,825.63,777.60,745.25,717.42,697.67,671.03;lc-ms c31h20n4o2(准确质量:480,97.79%)。
[0117]
向铝盘中加入14克邻苯二甲腈单体。然后将铝盘放入220℃烘箱中以熔化材料。随后,所述材料在如下条件下逐级固化:260℃6小时、300℃3小时和350℃4小时。也确定该固化产物的dma和tga。结果示于下表1和2中。
[0118]
实施例6:双酚c邻苯二甲腈(bisc-pn)的合成和均聚
[0119][0120]
向装有温度计、冷凝器和氮气入口的1000ml的四颈圆底烧瓶中加入双酚c(76.9g,0.27mol)、4-硝基邻苯二甲腈(94.7g,0.55mol)和dmf(250ml)。然后加入粉碎的k2co3(104.2g,0.82mol)。将所得混合物加热至80-90℃持续2-3小时。将混合物冷却至环境温度,
并倒入稀hcl中以形成固体。沉淀物通过过滤收集,用水洗涤至中性,之后用甲醇洗涤。真空干燥固体,作为黄色固体得到142g(99%)粗产物,在280nm下通过hplc分析确定纯度大于97%。由乙腈进一步重结晶得到浅棕色粉末产物(87.5%的收率),纯度大于99%。在10℃/min下通过dsc扫描确定熔点为198℃。1h nmr(ppm,dmso)8.126(d,2h),7.860(d,2h),7.43-7.50(m,6h),7.21-7.25(m,4h);
13
c nmr(ppm,dmso)160.191(2c),153.850(2c),139.172(1c),136 299(2c),135.697(2c),131.119(4c),123.343(2c),122.636(2c),119.849(4c),119.039(1c),116.734(2c),115.740(2c),115.254(2c),108.726(2c),41.246(2c),29.284(4c);ftir(cm-1)3066.78,3041.52,2228.20,1606.69,1590.35,1560.94,1499.85,1483.70,1416.10,1306.05,1295.19,1279.97,1246.74,1210.05,1169.39,1161.41,1154.56,1102.68,1089.03,1016.18,978.14,967.00,952.81,941.27,916.08,878.81,869.26,857.37,844.35,829.40,819.90,781.88,744.44,721.67,703.65,692.30,681.79;lc-ms:c32h14cl2n4o2,(准确质量:532,99.31%);
[0121]
向铝盘中加入14克邻苯二甲腈单体。然后将铝盘放入220℃烘箱中以熔化材料。随后,所述材料在如下条件下逐级固化:260℃6小时、300℃3小时和350℃4小时。也确定该固化产物的dma和tga。结果示于下表1和2中。
[0122]
实施例7:双酚m邻苯二甲腈(bism-pn)的合成和均聚
[0123][0124]
向装有温度计、冷凝器和氮气入口的1000ml的四颈圆底烧瓶中加入双酚m(120g,0.35mol)、4-硝基邻苯二甲腈(119.9g,0.69mol)和dmf(420ml)。然后加入粉碎的k2co3(105.6g,0.83mol)。将所得混合物加热至80-90℃持续2-3小时。将混合物冷却至环境温度,并倒入稀hcl中以形成固体。沉淀物通过过滤收集,用水洗涤至中性,之后用甲醇洗涤。真空干燥固体,作为深褐色固体得到205g(98%)粗产物,在280nm下通过hplc分析确定纯度大于96%。由乙腈进一步重结晶得到浅棕色粉末产物(收率78%),纯度大于98%。在10℃/min下通过dsc扫描确定熔点为135.12℃。1h nmr(ppm,dmso)8.066(d,2h),7.67(d,2h),7.20-7.35(m,7h),7.05-7.11(m,7h),1.638(s,12h);
13
c nmr(ppm,dmso)159.988(2c),150.192(2c),148.376(2c),146.734(2c),135.092(2c),127.485(4c),126.664(1c),122.913(1c),122.631(2c),121.147(2c),120.525(2c),118.561(4c),115.516(2c),114.665(2c),114.170(2c),106.860(2c),41.246(2c),29.284(4c);ftir(cm-1)3108.42,3074.20,3040.59,2967.77,2932.21,2870.46,2234.51,1799.58,1591.81,1563.28,1503.29,1488.99,1460.22,1449.76,1417.43,1404.26,1386.74,1364.16,1310.41,1285.60,1257.00,1248.90,1212.76,1174.34,1164.33,1122.44,1102.23,1085.13,1075.58,967.63,954.21,931.93,900.74,889.01,853.17,833.42,799.02,767.81,755.62,709.86,669.62;lc-ms:c40h30n4o2(准确质量:598,98.69%)。
[0125]
向铝盘中加入14克邻苯二甲腈单体。然后将铝盘放入160℃烘箱中以熔化材料。随后,所述材料在如下条件下逐级固化:260℃6小时、300℃3小时和350℃4小时。也确定该固化产物的dma和tga。结果示于下表1和2中。
[0126]
实施例8:双酚p邻苯二甲腈(bisp-pn)的合成和均聚
[0127][0128]
向装有温度计、冷凝器和氮气入口的1000ml的四颈圆底烧瓶中加入双酚p(120g,0.35mol)、4-硝基邻苯二甲腈(119.9g,0.69mol)和dmf(420ml)。然后加入粉碎的k2co3(105.6g,0.83mol)。将所得混合物加热至80-90℃持续2-3小时。将混合物冷却至环境温度,并倒入稀hcl中以形成固体。沉淀物通过过滤收集,用水洗涤至中性,之后用甲醇洗涤。真空干燥固体,作为绿色固体得到203g(97%)粗产物,在280nm下通过hplc分析确定纯度大于90%。由乙腈进一步重结晶得到棕褐色粉末产物(77%的收率),纯度约96%。在10℃/min下通过dsc扫描确定熔点为202℃。1h nmr(ppm,cdcl3中)7.72(dd,2h),7.25-7.35(m,8h),7.18(s,4h),6.97(d,4h),1.711(s,12h);
13
c nmr(ppm,cdcl3)161.887(2c),151.282(2c),148.938(2c),147.390(2c),135.388(2c),129.002(4c),126.422(4c),121.528(1c),121.355(1c),119.960(4c),117.495(2c),115,437(2c),115.054(2c),108.607(2c),42.424(2c),30.837(4c);ftir(cm-1)3084.57,3066.92,3042.90,2963.99,2373.24,2233.54,1915.82,1670.85,1590.88,1565.28,1502.28,1483.15,1425.21,1405.91,1393.05,1362.04,1303.82,1290.16,1280.14,1247.91,1211.87,1175.44,1156.94,1122.50,1087.47,1082.14,1014.40,954.30,890.11,879.46,855.92,831.87,767.27,719.31,699.82;lc-ms:c40h30n4o2(准确质量:598,96.34%);。
[0129]
向铝盘中加入14克邻苯二甲腈单体。然后将铝盘放入200℃烘箱中以熔化材料。随后,所述材料在如下条件下逐级固化:260℃6小时、300℃3小时和350℃4小时。也确定该固化产物的dma和tga。结果示于下表1和2中。
[0130]
实施例9:苯酚糠基胺苯并嗪(苯酚-fa)的合成和均聚
[0131][0132]
向配备有温度计、冷凝器和氮气入口的2000ml四颈圆底烧瓶中加入苯酚(300g,3.19mol)、低聚甲醛(200.9g,6.69mol)和甲苯(880ml)。加热混合物至60℃,然后向搅拌混合物中加入糠基胺(309.4.1g,3.19mol)。然后将所得混合物加热至80℃持续1小时,然后至100℃,以通过共沸过程脱除大部分水。进一步加热反应混合物至110℃以完成过程。过滤后,用3n氢氧化钠洗涤,然后用水洗涤并干燥。在真空下脱除甲苯后,产物在真空中在100℃下进一步干燥3小时,冷却后作为棕色固体得到646g(94.5%)产物。材料熔点范围在10℃/min下通过dsc扫描确定为54-56℃,和不经进一步纯化直接用于组合物中。通过gpc分析确定主要组分单体约73%、二聚物约11%,和所述单体具有如下谱图:1h nmr(ppm,cdcl3):7.40(d,1h),7.11(dd,1h),6.93(d,1h),6.87(dd,1h),6.80(d,1h),6.32(d,1h),6.23(d,1h),4.87(s,2h),4.00(s,2h),3.91(s,2h);
13
c nmr(ppm,cdcl3):154.0,151.7,142.6,127.8,127.6,120.8,119.7,116.5,110.2,108.9,81.8,49.6,48.2.lc-ms:236.1074(m+h
+
,calc.236.1075)。
[0133]
向铝盘中加入14克苯酚糠基胺苯并嗪。然后将铝盘放入80℃真空烘箱中熔化和
脱气1小时。脱气后,所述材料在如下条件下逐级固化:120℃2小时、150℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0134]
实施例10:4,4
′‑
硫代二苯酚苯胺苯并嗪(tdp-an)的合成和均聚
[0135][0136]
向配备有温度计、冷凝器和氮气入口的2000ml四颈圆底烧瓶中加入4,4
′‑
硫代二苯酚(66g,0.3mol)、低聚甲醛(38.1g,1.27mol)和甲苯(300ml)。然后逐滴加入苯胺(56.3g,0.6mol)。然后将反应混合物加热至80℃持续1小时,进一步加热至100℃,并通过共沸精馏收集水。实施14小时后,反应混合物冷却下来,通过旋转蒸发脱除甲苯,将残余物在80℃真空烘箱中干燥过夜,作为粘稠固体得到420g(94.5%)产物。材料熔点在10℃/min下通过dsc扫描确定为℃,和不经进一步纯化直接用于组合物中。通过gpc分析确定单体约52%、二聚物约18%、三聚物约11%、四聚物约6%,和所述单体具有如下谱图:1h nmr(ppm,cdcl3):7.0-7.3(m,12h),6.856(dd,2h),6.72(d,2h),5.432(s,4h),4.610(s,4h);
13
c nmr(ppm,cdcl3):153.552(2c),147.518(2c),130.746(1c),129.996(1c),129.046(6c),128.823(1c),128.116(1c),126.097(1c),122.408(1c),120.562(2c),117.304(6c),78.818(2c),43.629(2c)..lcms:453.1694(m+h
+
,calc.453.1637)。
[0137]
向铝盘中加入14克苯并嗪。然后将铝盘放入80℃真空烘箱中熔化和脱气1小时。脱气后,所述材料在如下条件下逐级固化:120℃2小时、150℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0138]
实施例11:4,4
′‑
二苯酚糠基胺苯并嗪(bp-fa)的合成和均聚
[0139][0140]
向配备有温度计、冷凝器和氮气入口的2000ml四颈圆底烧瓶中加入二苯酚(200g,1.07mol)、低聚甲醛(135.4g,4.51mol)、糠基胺(208.5g,2.15mol)和(720ml)。将混合物加热至100℃直到完成。冷却后,白色沉淀通过过滤收集,并用更多的二烷和乙醇洗涤。将产物在80℃真空烘箱中进一步干燥过夜,作为灰白色固体得到420g(94.5%)产物。材料熔点在10℃/min下通过dsc扫描确定为约℃,和不经进一步纯化直接用于组合物中。通过gpc分析确定主要组分为约88%,和其具有如下谱图:1h nmr(ppm,cdcl3):7.21(d,2h),7.30(dd,2h),7.09(dd,2h),6,83(d,2h),6.34(m,2h),6.23(m,2h),4.90(s,2h),4.04(s,2h),3.96(s,2h);
13
c nmr(ppm,cdcl3):153.2(2c),151.6(2c),142.6(2c),133.9(2c),126.0(2c),125.7(2c),119.8(2c),116.6(2c),110.3(2c),108.9(2c),82.0(2c),49.7(2c),48.3(2c).lcms:
[0141]
向铝盘中加入14克。然后将铝盘放入150℃真空烘箱中熔化和脱气1小时。脱气后,
所述材料在如下条件下逐级固化:150℃2小时、160℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0142]
实施例12-17:邻苯二甲腈单体/苯并嗪的共聚(苯酚-apa/tmbf-pn;苯酚-apa/ftmbf-pn;苯酚-apa/bisa-pn;苯酚-apa/bisc-pn;苯酚-apa/bism-pn;苯酚-apa/bisp-pn)
[0143]
向六个4盎司的玻璃罐中分别加入20克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa;实施例1)。然后将玻璃罐放入80℃烘箱中直到材料熔化,然后在搅拌下向玻璃罐中分别加入实施例3和7所述的6克化合物。偶尔搅拌所得混合物,直到所加材料均溶解在熔化的苯并嗪中,其中温度分别达到95℃(实施例16苯酚-apa/bism-pn,利用实施例7的bism-pn)、120℃(实施例12苯酚-apa/tmbf-pn,利用实施例3的tmbf-pn实施例3)、140℃(实施例13苯酚-apa/ftmbf-pn,利用实施例4的ftmbf-pn)、短时150℃(实施例17苯酚-apa/bisp-pn,利用实施例8的bisp-pn)和160℃(实施例14、15苯酚-apa/bisa-pn&苯酚-apa/bisc-pn,利用实施例5和6的biaa-pn&bisc-pn)。然后将约13克混合物转移至铝盘中。在85℃下脱气后,所述混合物在如下条件下逐级固化:120℃2小时、150℃2小时、180℃2小时和200℃2小时或160℃2小时、170℃2小时、180℃2小时和200℃2小时。确定新制备的样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃或260℃下进一步后固化3-4小时,和确定该固化产物的dma和tga。结果示于下表1和2中。
[0144]
实施例18:邻苯二甲腈单体/苯并嗪(苯酚-apa/ftmbf-pn)的共聚向4盎司的玻璃罐中加入12克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa;实施例1)。然后将玻璃罐放入80℃烘箱中直到材料熔化,然后在搅拌下向玻璃罐中加入2.4克由呋喃基-2,2
′
,6,6
′‑
四甲基双酚f制备的邻苯二甲腈(ftbmf-pn;实施例5)。将所得混合物逐渐加热至150℃并偶尔搅拌,直到所加材料均溶解在熔化的苯并嗪中。将约12.5克混合物转移入铝盘中。在65℃下脱气后,所述混合物在如下条件下逐级固化:120℃2小时、150℃2小时、180℃2小时和200℃ 2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0145]
实施例19:邻苯二甲腈单体/苯并嗪(苯酚-apa/ftmbf-pn)的共聚向4盎司的玻璃罐中加入10.8克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa,实施例1)。然后将玻璃罐放入80℃烘箱中直到材料熔化,然后在搅拌下向玻璃罐中加入5.38克邻苯二甲腈(ftmbf-pn,实施例5)。将所得混合物逐渐加热至150℃并偶尔搅拌,直到所加材料均溶解在熔化的苯并嗪中。将约12.5克混合物转移入铝盘中。在65℃下脱气后,所述混合物在如下条件下逐级固化:120℃2小时、150℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0146]
实施例20:邻苯二甲腈单体/苯并嗪(苯酚-apa/bism-pn)的共聚向4盎司的玻璃罐中加入20克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa,实施例1)。然后将玻璃罐放入80
℃烘箱中直到材料熔化,然后在搅拌下向玻璃罐中加入10克双酚m-邻苯二甲腈(bism-pn,实施例7),直到全部溶解(达到130℃)。将约12.5克混合物转移入铝盘中。所述混合物在如下条件下逐级固化:140℃2小时、180℃2小时和220℃3小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在260℃下进一步后固化4小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0147]
实施例21:邻苯二甲腈单体/苯并嗪(苯酚-apa/tmbf-pn)的共聚向4盎司的玻璃罐中加入21克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa,实施例1)。然后将玻璃罐放入80℃烘箱中直到材料熔化,然后在搅拌下向玻璃罐中加入9克2,2
′
,6,6
′‑
四甲基双酚f衍生的邻苯二甲腈(tmbf-pn,实施例3)。将所得混合物逐渐加热至150℃并偶尔搅拌,直到所加材料均溶解在熔化的苯并嗪中。将约12.5克混合物转移入铝盘中。所述混合物在如下条件下逐级固化:140℃1小时、160℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在260℃下进一步后固化4小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0148]
实施例22:邻苯二甲腈单体/苯并嗪(烯丙基ph-apa/bism-pn)的共聚
[0149]
向4盎司的玻璃罐中加入20克2-烯丙基苯酚3-氨基苯基乙炔苯并嗪(烯丙基ph-apa,实施例2)和6克双酚m-邻苯二甲腈(bism-pn,实施例7)。然后将玻璃罐放入80℃烘箱中。将烘箱逐渐加热至130℃,偶尔搅拌所得混合物,直到所加材料均溶解在熔化的苯并嗪中。将约12.5克混合物转移入铝盘中。在85℃下脱气后,所述混合物在如下条件下逐级固化:140℃1小时、160℃2小时、180℃2小时和220℃3小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在260℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0150]
实施例23:邻苯二甲腈单体/苯并嗪(烯丙基ph-apa/tmbf-pn)的共聚
[0151]
向4盎司的玻璃罐中加入20克2-烯丙基苯酚3-氨基苯基乙炔苯并嗪(烯丙基ph-apa,实施例2),然后在搅拌下向玻璃罐中加入6克tmbf-邻苯二甲腈(tmbf-pn,实施例3)。然后将玻璃罐放入80℃烘箱中。将烘箱逐渐加热至130℃,偶尔搅拌所得混合物直到所加材料均溶解在熔化的苯并嗪中。将约12.5克混合物转移入铝盘中。在85℃下脱气后,所述混合物在如下条件下逐级固化:140℃1小时、160℃2小时、180℃2小时和220℃3小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在260℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0152]
实施例24:邻苯二甲腈单体/苯并嗪(苯酚-apa/苯酚-fa/bism-pn)的共聚
[0153]
向4盎司的玻璃罐中加入10克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa,实施例1)和10克苯酚糠基胺苯并嗪(苯酚-fa,实施例9)。然后将玻璃罐放入90℃烘箱中直到材料熔化,然后在搅拌下向玻璃罐中加入6.0克由双酚m衍生的邻苯二甲腈(bism-pn,实施例7)。然后将温度提升至130℃。偶尔搅拌所得混合物,直到所加材料均溶解在熔化的苯并嗪中,在100℃下脱气后,所述混合物在如下条件下逐级固化:120℃2小时、150℃2小时、180℃
2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0154]
实施例25:邻苯二甲腈单体/苯并嗪(苯酚-apa/tdp-an/bism-pn)的共聚
[0155]
向4盎司的玻璃罐中加入20克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa;实施例1)和10克4,4
′‑
硫代二苯酚苯胺苯并嗪(tdp-an;实施例10)。然后将玻璃罐放入90℃烘箱中直到材料熔化,然后在搅拌下向玻璃罐中加入10克双酚m邻苯二甲腈(bism-pn,实施例7)。偶尔搅拌所得混合物直到所加材料均溶解在熔化的苯并嗪中。将约12.5克混合物转移入铝盘中。在100℃下脱气后,所述混合物在如下条件下逐级固化:120℃2小时、150℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0156]
实施例26:邻苯二甲腈单体/苯并嗪(苯酚-apa/mt35700/tmbf-pn)的共聚
[0157]
向4盎司的玻璃罐中加入16克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa,实施例1)。然后将玻璃罐放入110℃烘箱中熔化,和之后在搅拌下向玻璃罐中加入4.0克由2,2
′
,6,6
′‑
四甲基双酚f衍生的邻苯二甲腈(tmbf-pn,实施例3)。偶尔搅拌所得混合物直到所加材料均溶解在熔化的苯并嗪中。然后加入7g商购的araldite mt35700(mt35700,双酚f/苯酚苯胺苯并嗪树脂;内部获得),直到所有材料均熔化。将约12.5克混合物转移入铝盘中。所述混合物在如下条件下逐级固化:140℃2小时、160℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化4小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0158]
实施例27:邻苯二甲腈单体/苯并嗪(苯酚-apa/苯酚-fa/bp-fa/bism-pn)的共聚
[0159]
向4盎司的玻璃罐中加入5克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa,实施例1)和5克苯酚糠基胺苯并嗪(苯酚-fa,实施例9)。然后将玻璃罐放入90℃烘箱中直到材料熔化,然后在搅拌下向玻璃罐中加入3.0克由双酚m衍生的邻苯二甲腈(bism-pn,实施例7)。然后在搅拌下向玻璃罐中加入13克4,4
′‑
二苯酚糠基胺苯并嗪(bp-fa,实施例11)。偶尔搅拌所得混合物直到所加材料均溶解在熔化的苯并嗪中。将约12.5克混合物转移入铝盘中。在100℃下脱气后,所述混合物在如下条件下逐级固化:120℃2小时、150℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃下进一步后固化3小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0160]
实施例28:邻苯二甲腈单体/苯并嗪(苯酚-apa/tdp-an/ftmbf-pn)的共聚
[0161]
向4盎司的玻璃罐中加入20克苯并嗪和邻苯二甲腈的混合物(苯酚-apa/ftmbf-pn;实施例13)。然后将玻璃罐放入110℃烘箱中直到材料熔化,然后在搅拌下向玻璃罐中加
入7克4,4
′‑
硫代二苯酚苯胺胺苯并嗪(tdp-an,实施例10)。偶尔搅拌所得混合物直到所加材料均熔化。将约12.5克混合物转移入铝盘中。所述混合物在如下条件下逐级固化:140℃2小时、160℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃进一步后固化4小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0162]
实施例29:邻苯二甲腈单体/苯并嗪(苯酚-apa/ftmbf-pn/tmbf-pn)的共聚
[0163]
向4盎司的玻璃罐中加入20克前面实施例制备的混合物(苯酚-apa/ftmbf-pn,实施例13)。然后将玻璃罐放入90℃烘箱中熔化,然后在搅拌下向玻璃罐中加入4.0克由2,2
′
,6,6
′‑
四甲基双酚f衍生的邻苯二甲腈(tmbf-pn,实施例3)。将烘箱温度逐渐升高至150℃,并偶尔搅拌所得混合物,直到所加材料均溶解在熔化的苯并嗪中。将约12.5克混合物转移入铝盘中。所述混合物在如下条件下逐级固化:150℃1小时、160℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在260℃下进一步后固化4小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0164]
实施例30:邻苯二甲腈单体/苯并嗪(苯酚-apa/bism-pn/bisa-pn)的共聚
[0165]
向4盎司的玻璃罐中加入12克苯酚3-氨基苯基乙炔苯并嗪(苯酚-apa,实施例1)。然后将玻璃罐放入130℃烘箱中熔化,然后在搅拌下向玻璃罐中加入8.0克由双酚m衍生的邻苯二甲腈(bism-pn,实施例7)。偶尔搅拌所得混合物直到所加材料均溶解在熔化的苯并嗪中。然后搅拌加入2克双酚a衍生的邻苯二甲腈(bisa-pn,实施例5)直到溶解。将约12.5克混合物转移入铝盘中。所述混合物在如下条件下逐级固化:140℃2小时、160℃2小时、180℃2小时和200℃2小时。确定新制备样品的dsc和一半固化产物的dma、tga。另一半固化产物在250℃进一步后固化4小时,也确定该固化产物的dma和tga。结果示于下表1和2中。
[0166]
表1
[0167]
[0168][0169]
表2
[0170]
[0171]
[0172][0173]
虽然已经详细描述了本发明各种实施方案的制备和应用,应该理解的是本发明提供许多可应用的发明概念,这些概念可以在很多种具体内容中具体体现。这里讨论的具体实施方案只是本发明制备和应用的具体方法的示例,和不构成对本发明的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1