1,3,5-三嗪衍生物或其溶剂合物的晶体及其制造方法与流程
1.本发明涉及一种1,3,5-三嗪衍生物或其溶剂合物的晶体及含有它们的药物组合物。进而涉及一种该1,3,5-三嗪衍生物或其溶剂合物的晶体及含有它们的药物组合物的制造方法。
背景技术:
2.腺苷三磷酸(atp)作为细胞内的能量源或磷酸化基质为人所知。另一方面,也已知其亦作为细胞外的信息传导物质发挥作用。进而,已知atp会因细胞的损伤、炎症、伤害刺激、血中氧浓度的降低等各种刺激而向细胞外释放、与其他神经传导物质一起自一级感觉神经末梢向细胞外释放。向细胞外释放的atp会经由atp受体进行各种细胞外信息传导(非专利文献4、非专利文献5)。
3.atp受体大致分为离子通道型的p2x家族及g蛋白偶联型的p2y家族。p2x受体家族中报告有7种亚型,形成同型三聚物或与其他p2x亚型的异型三聚物,作为非选择性阳离子通道发挥作用(非专利文献6)。
4.已知,atp会引起疼痛,进而根据使用p2x3的剔除或减弱(knock down)技术的研究,显示出p2x3受体会参与慢性疼痛的传导。p2x3受体于末梢感觉神经特异性地表达,形成同型复合体或与p2x2的异型复合体(p2x
2/3
)。(非专利文献1)
5.其后,提示了具有p2x3或p2x
2/3
受体拮抗作用的化合物于疼痛的治疗(专利文献1、非专利文献3及非专利文献7)、伴有排尿功能异常的疾病的治疗(非专利文献2)、呼吸器疾病的治疗(非专利文献8、非专利文献9、非专利文献10、专利文献2及专利文献3)、慢性咳嗽的治疗(专利文献4、专利文献5及非专利文献11)、高血压的治疗(非专利文献12)、胰脏炎所伴随的疼痛的治疗(非专利文献13)、子宫内膜症所伴随的疼痛的治疗(非专利文献14及非专利文献15)中可能有用。
6.继而,专利文献6中记载有:下式:[化学式1]所表示的1,3,5-三嗪衍生物具有p2x3和/或p2x
2/3
拮抗作用,对疼痛的治疗和/或预防有用。
[0007]
专利文献7中记载有:下式:[化学式2]
所表示的1,3,5-三嗪衍生物具有p2x3和/或p2x
2/3
拮抗作用,对疼痛的治疗和/或预防有用。
[0008]
专利文献8中记载有:下式:[化学式3]所表示的1,3,5-三嗪衍生物具有p2x3和/或p2x
2/3
拮抗作用,对疼痛的治疗和/或预防有用。专利文献9中记载有:下式:[化学式4]所表示的1,3,5-三嗪衍生物具有p2x3和/或p2x
2/3
拮抗作用,对疼痛的治疗和/或预防有用。另外,专利文献9的实施例中公开有以下的化合物(i-127):[化学式5]
但未公开该化合物的晶体。此外,专利文献6、7、8及9中公开有1,3,5-三嗪衍生物的制造方法,但并未记载本发明的制造方法,仅公开了类似化合物的制造方法。另外,专利文献10中公开了对慢性咳嗽显示出治疗效果的1,3,5-三嗪衍生物,但并未记载本发明的晶体及制造方法。另外,非专利文献16中公开了(s)-1-苯基乙基胺与甲基丙烯酸甲酯的氮杂-迈克尔加成反应。背景技术文献专利文献
[0009]
专利文献1:国际公开第02/094767号说明书专利文献2:国际公开第2006/012639号说明书专利文献3:国际公开第2010/149578号说明书专利文献4:国际公开第2015/027212号说明书专利文献5:国际公开第2017/058645号说明书专利文献6:国际公开第2010/092966号说明书专利文献7:国际公开第2012/020749号说明书专利文献8:国际公开第2013/089212号说明书专利文献9:国际公开第2014/200078号说明书专利文献10:国际公开第2020/071530号说明书非专利文献
[0010]
非专利文献1:neuroscientist 2005年,第11卷,p.345-356非专利文献2:j.physiol.567.2 2005年p.621-639非专利文献3:expert opin.ther.patens 2006年16卷,8号,p.1113-1127非专利文献4:j.physiology 2003年,554卷,2号,p.301-308非专利文献5:j.physiology 2003年,553卷,3号,p.683-694非专利文献6:pflungers arch eur j physiol 2006年,p.452,513-537非专利文献7:pnas 2002年,99卷,26号,p.17179-17184非专利文献8:brouns et al.am j respir cell moi biol 2000年,第23卷,p.52-61非专利文献9:basoglu et al.chest.2005年,第128卷,4号,p.1905-9非专利文献10:adriaensen et al.the anatomical record part a 2003年,第270a卷,p.25-40
非专利文献11:lancet,2015年,第385卷,p.1198-205非专利文献12:nat med 2016年,第22卷,p.1151-1159非专利文献13:am j physiol gastrointest liver physiol 2015年,308卷,p710-719非专利文献14:plos one 2017年,12卷,9号非专利文献15:international journal of nanomedicine 2017年,12卷,8171-8183非专利文献16:tetrahedron asymmetry,vol.7,no.3,pp.699-708,1996
技术实现要素:
发明所要解决的问题
[0011]
药物活性成分根据各自的固体形态可具有实质上不同的物理特性。这样的物理特性的不同例如可对药物活性成分的制造方法或者投予方法、或包含药物活性成分的药物组合物产生影响。本发明提供一种1,3,5-三嗪衍生物或其溶剂合物的晶体,其与其他固体形态相比,于药物活性成分的制造方法或者投予方法、或包含药物活性成分的药物组合物中非常有用。一般而言,作为药品有用的化合物的晶体的物性会对药物的生物利用度、原料药的纯度、制剂的配方等产生较大影响,因此于药品开发中极其重要。因此,关于式(i)所示的化合物,需要研究什么样的晶体形式作为药品最优异。即,所述物性取决于各化合物的属性,因此一般而言,难以预测具有良好的物性的原料药用的晶体形式,要求针对各化合物实际进行各种研究。因此,本发明的课题在于针对式(i)所示的化合物提供一种作为原料药具有良好的物性的晶体形式。另外,专利文献9中未记载化合物i-127的制造方法,但作为类似的化合物,专利文献9的参考例3中公开有如下式所示的1,3,5-三嗪衍生物的制造方法。然而,该制造方法尚不能说是充分的,存在改良的余地。[化学式6]解决问题的技术手段
[0012]
本发明人等进行深入研究的结果,发现在式(i)所示的化合物中存在无水物i型、无水物ii型、二水合物的晶体形式。进而发现,无水物i型晶体及二水合物晶体与其他晶体形式相比更稳定。并且发现,无水物i型晶体与其他晶体形式相比,晶体的压缩度(%)较低,晶体的流动性良好。另外,本发明人等发现了化学纯度和/或光学纯度较高的中间体及它们的制造方法、以及具有p2x3和/或p2x
2/3
拮抗作用的光学活性的1,3,5-三嗪衍生物的制造方法。本发明涉及以下的项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')至
(35')、(3”)、(5”)及(36”)至(42”)。(1')一种式(i)所示的化合物或其溶剂合物的晶体:[化学式7](2')如上述项目(1')记载的化合物的无水物i型晶体,其于粉末x射线衍射光谱中,于衍射角度(2θ):15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
及25.4
°±
0.2
°
、或衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
及19.4
°±
0.2
°
处具有特征峰。(2'a)如上述项目(1')记载的化合物的无水物i型晶体,其于粉末x射线衍射光谱中,于衍射角度(2θ):15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
及25.4
°±
0.2
°
处具有特征峰。(2'b)如上述项目(1')记载的化合物的无水物i型晶体,其于粉末x射线衍射光谱中,于衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
及19.4
°±
0.2
°
处具有特征峰。(3')如上述项目(1')记载的化合物的无水物i型晶体,其于粉末x射线衍射光谱中,于衍射角度(2θ):12.6
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
、26.6
°±
0.2
°
、27.8
°±
0.2
°
及32.8
°±
0.2
°
、或衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、17.2
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
及27.8
°±
0.2
°
处具有特征峰。(3'a)如上述项目(1')记载的化合物的无水物i型晶体,其于粉末x射线衍射光谱中,于衍射角度(2θ):12.6
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
、26.6
°±
0.2
°
、27.8
°±
0.2
°
及32.8
°±
0.2
°
处具有特征峰。(3'b)如上述项目(1')记载的化合物的无水物i型晶体,其于粉末x射线衍射光谱中,于衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、17.2
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
及27.8
°±
0.2
°
处具有特征峰。(3”)如上述项目(1')记载的化合物的无水物i型晶体,其于拉曼光谱中,于829cm-1
±
2cm-1
、989cm-1
±
2cm-1
、1013cm-1
±
2cm-1
、1128cm-1
±
2cm-1
及1370cm-1
±
2cm-1
处具有吸收峰。
[0013]
(4')如上述项目(1')记载的化合物的二水合物晶体,其于粉末x射线衍射光谱中,
于衍射角度(2θ):5.7
°±
0.2
°
、7.7
°±
0.2
°
、11.8
°±
0.2
°
、15.2
°±
0.2
°
及17.7
°±
0.2
°
处具有特征峰。(5')如上述项目(1')记载的化合物的二水合物晶体,其于粉末x射线衍射光谱中,于衍射角度(2θ):5.7
°±
0.2
°
、7.7
°±
0.2
°
、11.8
°±
0.2
°
、15.2
°±
0.2
°
、17.7
°±
0.2
°
、20.6
°±
0.2
°
、20.8
°±
0.2
°
、26.5
°±
0.2
°
、27.1
°±
0.2
°
及29.1
°±
0.2
°
处具有特征峰。(5”)如上述项目(1')记载的化合物的二水合物晶体,其于拉曼光谱中,于871cm-1
±
2cm-1
、996cm-1
±
2cm-1
、1114cm-1
±
2cm-1
、1234cm-1
±
2cm-1
、1340cm-1
±
2cm-1
及1577cm-1
±
2cm-1
处具有吸收峰。
[0014]
(6')一种包含如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体的药物组合物。(7')一种如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体的制造方法。(8')如上述项目(6')记载的药物组合物,其为p2x3和/或p2x
2/3
拮抗剂。(9')如上述项目(6')记载的药物组合物,其用于慢性咳嗽的治疗和/或预防。(10')如上述项目(6')记载的药物组合物,其用于难治性慢性咳嗽的治疗和/或预防。(11')一种p2x3和/或p2x
2/3
拮抗剂,其特征在于含有如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体。(12')一种慢性咳嗽的治疗和/或预防剂,其特征在于含有如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体。(13')一种难治性慢性咳嗽的治疗和/或预防剂,其特征在于含有如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体。(14')一种慢性咳嗽的治疗和/或预防方法,其特征在于投予包含如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体的药物组合物。(15')一种难治性慢性咳嗽的治疗和/或预防方法,其特征在于投予包含如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体的药物组合物。(16')如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体在用于制造用于治疗和/或预防慢性咳嗽的药物中的用途。(17')如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体在用于制造用于治疗和/或预防难治性慢性咳嗽的药物中的用途。(18')如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体,其用于治疗和/或预防慢性咳嗽。(19')如上述项目(1')、(2')、(2'a)、(2'b)、(3')、(3'a)、(3'b)、(4')、(5')、(3”)及(5”)中任一项记载的晶体,其用于治疗和/或预防难治性慢性咳嗽。(20')如上述项目(1')记载的晶体,其通过与图1实质上一致的粉末x射线衍射光
谱赋予特征。(21')如上述项目(2')记载的晶体,其通过与图1实质上一致的粉末x射线衍射光谱赋予特征。(22')如上述项目(2'a)记载的晶体,其通过与图1实质上一致的粉末x射线衍射光谱赋予特征。(23')如上述项目(2'b)记载的晶体,其通过与图1实质上一致的粉末x射线衍射光谱赋予特征。(24')如上述项目(3')记载的晶体,其通过与图1实质上一致的粉末x射线衍射光谱赋予特征。(25')如上述项目(3'a)记载的晶体,其通过与图1实质上一致的粉末x射线衍射光谱赋予特征。(26')如上述项目(3'b)记载的晶体,其通过与图1实质上一致的粉末x射线衍射光谱赋予特征。(27')如上述项目(4')记载的晶体,其通过与图4实质上一致的粉末x射线衍射光谱赋予特征。(28')如上述项目(5')记载的晶体,其通过与图4实质上一致的粉末x射线衍射光谱赋予特征。(29')如上述项目(1')记载的晶体,其通过与图2实质上一致的拉曼光谱赋予特征。(30')如上述项目(1')记载的化合物的无水物i型晶体,其通过选自由以下的(i)及(ii)所组成的组的1种以上物理化学性质赋予特征:(i)于粉末x射线衍射光谱中,于衍射角度(2θ):15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
及25.4
°±
0.2
°
、或衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
及19.4
°±
0.2
°
处具有特征峰;(ii)于拉曼光谱中,于829cm-1
±
2cm-1
、989cm-1
±
2cm-1
、1013cm-1
±
2cm-1
、1128cm-1
±
2cm-1
及1370cm-1
±
2cm-1
处具有吸收峰。(31')如上述项目(1')记载的化合物的无水物i型晶体,其通过选自由以下的(i)及(ii)所组成的组的1种以上物理化学性质赋予特征:(i)于粉末x射线衍射光谱中,于衍射角度(2θ):15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
及25.4
°±
0.2
°
处具有特征峰;(ii)于拉曼光谱中,于829cm-1
±
2cm-1
、989cm-1
±
2cm-1
、1013cm-1
±
2cm-1
、1128cm-1
±
2cm-1
及1370cm-1
±
2cm-1
处具有吸收峰。(32')如上述项目(1')记载的化合物的无水物i型晶体,其通过选自由以下的(i)及(ii)所组成的组的1种以上物理化学性质赋予特征:(i)于粉末x射线衍射光谱中,于衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
及19.4
°±
0.2
°
处具有特征峰;(ii)于拉曼光谱中,于829cm-1
±
2cm-1
、989cm-1
±
2cm-1
、1013cm-1
±
2cm-1
、1128cm-1
±
2cm-1
及1370cm-1
±
2cm-1
处具有吸收峰。
(33')如上述项目(1')记载的化合物的无水物i型晶体,其通过选自由以下的(i)及(ii)所组成的组的1种以上物理化学性质赋予特征:(i)于粉末x射线衍射光谱中,于衍射角度(2θ):12.6
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
、26.6
°±
0.2
°
、27.8
°±
0.2
°
及32.8
°±
0.2
°
、或衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、17.2
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
及27.8
°±
0.2
°
处具有特征峰;(ii)于拉曼光谱中,于829cm-1
±
2cm-1
、989cm-1
±
2cm-1
、1013cm-1
±
2cm-1
、1128cm-1
±
2cm-1
及1370cm-1
±
2cm-1
处具有吸收峰。(34')如上述项目(1')记载的化合物的无水物i型晶体,其通过选自由以下的(i)及(ii)所组成的组的1种以上物理化学性质赋予特征:(i)于粉末x射线衍射光谱中,于衍射角度(2θ):12.6
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
、26.6
°±
0.2
°
、27.8
°±
0.2
°
及32.8
°±
0.2
°
处具有特征峰;(ii)于拉曼光谱中,于829cm-1
±
2cm-1
、989cm-1
±
2cm-1
、1013cm-1
±
2cm-1
、1128cm-1
±
2cm-1
及1370cm-1
±
2cm-1
处具有吸收峰。(35')如上述项目(1')记载的化合物的无水物i型晶体,其通过选自由以下的(i)及(ii)所组成的组的1种以上物理化学性质赋予特征:(i)于粉末x射线衍射光谱中,于衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、17.2
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
及27.8
°±
0.2
°
处具有特征峰;(ii)于拉曼光谱中,于829cm-1
±
2cm-1
、989cm-1
±
2cm-1
、1013cm-1
±
2cm-1
、1128cm-1
±
2cm-1
及1370cm-1
±
2cm-1
处具有吸收峰。(36”)如上述项目(1')记载的化合物的无水物i型晶体,其通过选自由以下的(a)及(b)所组成的组的1种以上光谱和/或曲线赋予特征:(a)与图1实质上一致的粉末x射线衍射光谱;(b)与图2实质上一致的拉曼光谱。
[0015]
(37”)如上述项目(1')记载的晶体,其通过与图6实质上一致的拉曼光谱赋予特征。(38”)如上述项目(1')记载的化合物的二水合物晶体,其通过选自由以下的(i)及(ii)所组成的组的1种以上物理化学性质赋予特征:(i)于粉末x射线衍射光谱中,于衍射角度(2θ):5.7
°±
0.2
°
、7.7
°±
0.2
°
、11.8
°±
0.2
°
、15.2
°±
0.2
°
及17.7
°±
0.2
°
处具有特征峰;(ii)于拉曼光谱中,于871cm-1
±
2cm-1
、996cm-1
±
2cm-1
、1114cm-1
±
2cm-1
、1234cm-1
±
2cm-1
、1340cm-1
±
2cm-1
及1577cm-1
±
2cm-1
处具有吸收峰。(39”)如上述项目(1')记载的化合物的二水合物晶体,其通过选自由以下的(i)及(ii)所组成的组的1种以上物理化学性质赋予特征:(i)于粉末x射线衍射光谱中,于衍射角度(2θ):5.7
°±
0.2
°
、7.7
°±
0.2
°
、11.8
°±
0.2
°
、15.2
°±
0.2
°
、17.7
°±
0.2
°
、20.6
°±
0.2
°
、20.8
°±
0.2
°
、26.5
°±
0.2
°
、27.1
°±
0.2
°
及29.1
°±
0.2
°
处具有特征峰;(ii)于拉曼光谱中,于871cm-1
±
2cm-1
、996cm-1
±
2cm-1
、1114cm-1
±
2cm-1
、1234cm-1
±
2cm-1
、1340cm-1
±
2cm-1
及1577cm-1
±
2cm-1
处具有吸收峰。(40”)如上述项目(1')记载的化合物的二水合物晶体,其通过选自由以下的(a)及(b)所组成的组的1种以上光谱和/或曲线赋予特征:(a)与图4实质上一致的粉末x射线衍射光谱;(b)与图6实质上一致的拉曼光谱。(41”)如上述项目(1')记载的化合物的无水物i型晶体,其于以298.15k进行测定的情形时,与以下的晶体学资料实质上一致:空间群:p1空间群:p1空间群:p1α=67.712(4)
°
β=80.870(4)
°
γ=80.870(4)
°
。(42”)如上述项目(1')记载的化合物的无水物i型晶体,其于以298.15k进行测定的情形时,通过以下的晶体学数据赋予特征:空间群:p1空间群:p1空间群:p1α=67.7
°±
0.5
°
β=80.9
°±
0.5
°
γ=86.9
°±
0.5
°
。
[0016]
另外,本发明涉及以下的项目(1)至(34)。(1)一种式(i)所示的化合物或其溶剂合物的晶体:[化学式8]
(2)如上述项目(1)记载的化合物的晶体,其于粉末x射线衍射光谱中,于衍射角度(2θ):15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
或7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、19.4
°±
0.2
°
处具有特征峰。(3)如上述项目(1)记载的化合物的晶体,其于粉末x射线衍射光谱中,于衍射角度(2θ):12.6
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
、26.6
°±
0.2
°
、27.8
°±
0.2
°
、32.8
°±
0.2
°
或7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、17.2
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
、27.8
°±
0.2
°
处具有特征峰。(4)一种包含如上述项目(1)至(3)中任一项记载的晶体的药物组合物。(5)一种如上述项目(1)至(3)中任一项记载的晶体的制造方法。(6)一种式(iv)所示的化合物或其盐的制造方法:[化学式11](式中,r1为c1-c4烷基);其特征在于:使式(ii):[化学式9]所表示的化合物或其盐与式(iii):[化学式10](式中,r1为c1-c4烷基)所示的化合物或其盐,在选自由氯化锂、氯化钙、氯化镁、溴化锂、对甲苯磺酸、甲磺酸及三氟甲磺酸所组成的组的1种以上添加剂的存在下反应。(7)如上述项目(6)记载的制造方法,其中添加剂为氯化锂。(8)一种式(iv-a)所示的化合物的对甲苯磺酸盐的制造方法:[化学式12](式中,r1为c1-c4烷基);其特征在于:通过如上述项目(6)或(7)记载的制造方法获得式(iv)所示的化合物或其盐,并添加对甲苯磺酸。(9)一种式(iv-a)所示的化合物的对甲苯磺酸盐的制造方法:[化学式13]
(式中,r1为c1-c4烷基);其特征在于:于如上述项目(6)记载的制造方法中,添加剂为对甲苯磺酸。(10)如上述项目(6)至(9)中任一项记载的制造方法,其中r1为甲基。(11)一种式(iv-b)所示的化合物的对甲苯磺酸盐:[化学式14](12)一种式(v)所示的化合物的1/2硫酸盐的制造方法:[化学式16]其特征在于:对式(iv-b):[化学式15]所表示的化合物的对甲苯磺酸盐进行氢化分解反应,并添加硫酸。(13)一种式(v)所示化合物的1/2硫酸盐:[化学式17](14)一种式(i)所示的化合物或其盐的制造方法:[化学式19]其特征在于:将式(vi):
[化学式18](式中,r1为c1-c4烷基)所示的化合物或其盐,在选自由异丙醇、四氢呋喃及叔丁醇所组成的组的1种以上溶剂的存在下进行水解反应。(15)如上述项目(14)记载的制造方法,其中r1为甲基。(16)一种式(vi)所示的化合物或其盐的制造方法:[化学式21](式中,r1为甲基);该方法包括:通过如上述项目(6)至(10)中任一项记载的方法制造式(iv-b):[化学式20]所表示的化合物的对甲苯磺酸盐的步骤。(17)一种式(vi)所示的化合物或其盐的制造方法:[化学式23](式中,r1为甲基);该方法包括:通过如上述项目(12)记载的方法制造式(v):[化学式22]
所表示的化合物的1/2硫酸盐的步骤。(18)一种式(vi)所示的化合物或其盐的制造方法:[化学式26](式中,r1为甲基);该方法包括:通过如上述项目(6)至(10)中任一项记载的方法制造式(iv-b):[化学式24]所表示的化合物的对甲苯磺酸盐的步骤;及通过如上述项目(12)记载的方法制造式(v)的步骤:[化学式25]所表示的化合物的1/2硫酸盐。(19)一种(i)所示的化合物或其盐的制造方法:[化学式28]其特征在于:通过如上述项目(16)至(18)中任一项记载的制造方法获得式(vi):[化学式27]
(式中,r1为甲基)所示的化合物或其盐,并将所获得的式(vi)所示的化合物或其盐在选自由异丙醇、四氢呋喃及叔丁醇所组成的组的1种以上溶剂的存在下进行水解反应。(20)如上述项目(4)记载的药物组合物,其为p2x3和/或p2x
2/3
拮抗剂。(21)如上述项目(4)记载的药物组合物,其用于慢性咳嗽的治疗和/或预防。(22)如上述项目(4)记载的药物组合物,其用于难治性慢性咳嗽的治疗和/或预防。(23)一种p2x3和/或p2x
2/3
拮抗剂,其特征在于含有如上述项目(1)至(3)中任一项记载的晶体。(24)一种慢性咳嗽的治疗和/或预防剂,其特征在于含有如上述项目(1)至(3)中任一项记载的晶体。(25)一种难治性慢性咳嗽的治疗和/或预防剂,其特征在于含有如上述项目(1)至(3)中任一项记载的晶体。(26)一种慢性咳嗽的治疗和/或预防方法,其特征在于投予包含如上述项目(1)至(3)中任一项记载的晶体的药物组合物。(27)一种难治性慢性咳嗽的治疗和/或预防方法,其特征在于投予包含如上述项目(1)至(3)中任一项记载的晶体的药物组合物。(28)如上述项目(1)至(3)中任一项记载的晶体在用于制造用于治疗和/或预防慢性咳嗽的药物中的用途。(29)如上述项目(1)至(3)中任一项记载的晶体在用于制造用于治疗和/或预防难治性慢性咳嗽的药物中的用途。(30)如上述项目(1)至(3)中任一项记载的晶体,其用于慢性咳嗽的治疗和/或预防。(31)如上述项目(1)至(3)中任一项记载的晶体,其用于难治性慢性咳嗽的治疗和/或预防。(32)如上述项目(1)记载的晶体,其通过与图1实质上一致的粉末x射线衍射光谱赋予特征。(33)如上述项目(2)记载的晶体,其通过与图1实质上一致的粉末x射线衍射光谱赋予特征。(34)如上述项目(3)记载的晶体,其通过与图1实质上一致的粉末x射线衍射光谱赋予特征。(35)如上述项目(1)记载的晶体,其通过与图2实质上一致的拉曼光谱赋予特征。(36)如上述项目(2)记载的晶体,其通过与图2实质上一致的拉曼光谱赋予特征。
magnetic resonance,核磁共振)。横轴表示化学位移(δ)值,纵轴表示质子信号的相对强度。图10示出式(i)所示的化合物的乙酸乙酯/己烷溶剂合物晶体的粉末x射线衍射图案。横轴为2θ(
°
),纵轴表示强度(count)。图11示出式(i)所示的化合物的乙酸乙酯/己烷溶剂合物晶体的拉曼光谱。横轴表示拉曼位移(cm-1
),纵轴表示峰强度。图12示出式(i)所示的化合物的无水物ii型晶体(form ii)的粉末x射线衍射图案。横轴为2θ(
°
),纵轴表示强度(count)。图13示出式(i)所示的化合物的乙酸乙酯/己烷溶剂合物晶体的tg/dta分析结果。纵轴表示热量(μv)或重量变化(%),横轴表示温度(℃)。图中的cel意指摄氏度(℃)。
具体实施方式
[0019]
固体形态的选择及控制对于特别是成为药物的化合物较重要。固体形态的慎重选择及控制能够减少与化合物相关的制造、配制或投予的问题。
[0020]
只要未特别提及,则本说明书中及权利要求书记载的数值为大致的值。数值的变动起因于装置校准、装置误差、物质的纯度、晶体大小、样品大小、温度、及其他因素。
[0021]
本说明书中所使用的“晶体”意指构成的原子、离子、分子等三维地规律地排列而成的固体,与不具有这样的规律的内部结构的非晶质固体相区别。本发明的晶体可为单晶、双晶、多晶等。进而,“晶体”中有时存在多晶型,包含它们在内称为“晶体形态”,包含于本发明中。除此以外,“式(i)所示的化合物”可形成与水的溶剂合物(即,水合物)或与普通有机溶剂的溶剂合物,这样的溶剂合物也包含于本发明的范围内。晶体形态及结晶度例如可通过包括粉末x射线衍射测定、拉曼光谱法、红外吸收光谱测定法、水分吸附脱附测定、示差扫描量热测定、及溶解特性的诸多技术进行测定。
[0022]
本说明书中所使用的“盐”意指例如“式(i)所示的化合物”与反向分子于同一晶格内规律地排列,且可包含任意数量的反向分子。是指于晶格中于化合物与反向分子之间通过质子转移而介导离子键者。
[0023]
盐形成的研究提供在不改变其化学结构的情况下改变药剂的物理化学特征及所获得的生物学特征的方法。盐的形成可对药剂的特性产生很大影响。在选择适当的盐时,盐的吸湿性、稳定性、溶解度及加工特性亦为重要的观点。盐的溶解度可能会影响其作为药剂使用的适性。在水性溶解度较低的情形下,生体内(in vivo)投予时的溶解速度在吸收过程中会受到速率限制,可能会带来较低的生物利用度。另外,由于水性溶解度较低,通过注射进行投予可能变得困难,因此在适当的投予途径的选择上可能产生限制。
[0024]
上述“式(i)所示的化合物”可转换为溶剂合物、药学上可接受的盐、或盐的溶剂合物。在本发明的一个方面中,本化合物为碱加成盐的形式。碱加成盐包含由包含无机及有机碱的药学上可接受的非毒性碱所制造的盐。来自无机碱的盐包含铝、钙、锂、钾、镁、钠、锌及其他金属盐,但并不限定于此。来自药学上可接受的非毒性碱的盐包含一级、二级或三级胺类、天然存在的取代胺类、环状胺类及碱性离子交换树脂、例如精氨酸、甜菜碱、苄星、咖啡
因、胆碱、氯普鲁卡因、环普鲁卡因、n'n'-二苄基乙二胺、二乙醇胺、二乙胺、2-二乙基-氨基乙醇、2-二甲基氨基乙醇、乙醇胺、乙二胺、n-乙基-吗啉、n-乙基哌啶、还原葡糖胺、葡萄糖胺、组氨酸、海卓胺、异丙基胺、赖氨酸、葡甲胺、吗啉、哌嗪、哌啶、聚胺树脂、普鲁卡因、嘌呤类、三级丁胺(2-甲基丙烷-2-胺)、可可碱、包含三乙胺、三甲胺、三丙胺、胺丁三醇等的取代胺类、以及非毒性铵及四级铵、及包含铵、四甲基铵、四乙基铵但不限定于所述的阳离子的盐。
[0025]“式(i)所示的化合物”、“式(ii)所示的化合物”、“式(iv)所示的化合物”、“式(iv-a)所示的化合物”、“式(iv-b)所示的化合物”及“式(v)所示的化合物”的酸加成盐的例中,包含具有无机酸例如盐酸、氢溴酸、正磷酸、硝酸、磷酸或硫酸等、或有机酸例如甲酸、甲磺酸、乙磺酸、对甲苯磺酸、乙酸、丙酸、乳酸、柠檬酸、反丁烯二酸、苹果酸、琥珀酸、水杨酸、顺丁烯二酸、甘油磷酸、酒石酸、苯甲酸、谷氨酸、天冬氨酸、苯磺酸、2-萘磺酸等萘磺酸、己酸或乙酰水杨酸等者。
[0026]
本说明书中所使用的“溶剂合物”例如指相对于“式(i)所示的化合物”与任意数量的溶剂分子规律地排列者。作为溶剂分子,例如可例举:乙腈、氯苯、氯仿、环己烷、1,2-二氯乙烯、二氯甲烷、1,2-二甲氧基乙烷、n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、1,4-二噁烷、2-乙氧基乙醇、乙二醇、甲酰胺、己烷、甲醇、2-甲氧基乙醇、甲基丁基酮、甲基环己烷、n-甲基吡咯烷酮、硝基甲烷、吡啶、环丁砜、萘满、甲苯、1,1,2-三氯乙烯、二甲苯、乙酸、苯甲醚、1-丁醇、2-丁醇、叔丁醇、乙酸正丁酯、叔丁基甲醚、异丙苯、二甲基亚砜、乙酸乙酯、二乙醚、甲酸乙酯、甲酸、庚烷、乙酸异丁酯、乙酸异丙酯、乙酸甲酯、3-甲基-1-丁醇、甲基乙基酮、甲基异丁基酮、2-甲基-1-丙醇、戊烷、1-戊醇、1-丙醇、2-丙醇、乙酸丙酯、四氢呋喃、水(即水合物)、乙醇、丙酮、1,1-二乙氧基丙烷、1,1-二甲氧基甲烷、2,2-二甲氧基丙烷、异辛烷、异丙醚、甲基异丙基酮、甲基四氢呋喃、石油醚、三氯乙酸及三氟乙酸。优选可例举:乙酸、苯甲醚、1-丁醇、2-丁醇、乙酸正丁酯、叔丁基甲醚、异丙苯、二甲基亚砜、乙酸乙酯、二乙醚、甲酸乙酯、甲酸、庚烷、乙酸异丁酯、乙酸异丙酯、乙酸甲酯、3-甲基-1-丁醇、甲基乙基酮、甲基异丁基酮、2-甲基-1-丙醇、戊烷、1-戊醇、1-丙醇、2-丙醇、乙酸丙酯、四氢呋喃、水(即水合物)、乙醇、丙酮、1,1-二乙氧基丙烷、1,1-二甲氧基甲烷、2,2-二甲氧基丙烷、异辛烷、异丙醚、甲基异丙基酮、甲基四氢呋喃、石油醚、三氯乙酸及三氟乙酸。更优选可例举:水(即水合物)、乙醇、丙酮、1,1-二乙氧基丙烷、1,1-二甲氧基甲烷、2,2-二甲氧基丙烷、异辛烷、异丙醚、甲基异丙基酮、甲基四氢呋喃、石油醚、三氯乙酸及三氟乙酸等。另外,通过将“式(i)所示的化合物”放置于大气中,会吸收水分,存在附着吸附水的情况、或形成水合物的情况。关于“式(ii)所示的化合物”、“式(iii)所示的化合物”、“式(iv)所示的化合物”、“式(iv-a)所示的化合物”、“式(iv-b)所示的化合物”、“式(v)所示的化合物”及“式(vi)所示的化合物”,亦可形成溶剂合物。
[0027]
本发明的水合物或其晶体例如相对于“式(i)所示的化合物”含有约2摩尔当量的水分子。作为本发明的水合物晶体,优选可例举二水合物。
本发明的水合物或其晶体的水分含量例如可例举4.7~9.7重量%。优选为可例举约5.6~7.6重量%(二水合物的理论值为6.6%,但由于附着于晶体的水的影响,亦存在水分含量变高的情况、或于测定前晶体中的水的一部分脱附而使水分含量变低的情况)。
[0028]
本发明的晶体亦可为氘转换体。本发明的晶体亦可利用同位素(例如3h、
14
c、
35
s、
125
i等)进行标记。
[0029]
本说明书中所使用的“无水物”与“无溶剂合物”、“非溶剂合物”、“无水合物”及“非水合物”同义。
[0030]
式(i):[化学式29]所表示的化合物为专利文献9中所记载的p2x3和/或p2x
2/3
拮抗剂。作为慢性咳嗽的治疗剂或预防剂非常有用。式(i)所示的化合物可参考本技术实施方案进行制备。
[0031]
式(i)所示的化合物的互变异构体为式(i'):[化学式30]所示的化合物(氨基体),与式(i)所示的化合物同样地具有p2x3和/或p2x
2/3
受体拮抗的作用。另外,式(vi)所示的化合物亦可与上述同样地地采用互变异构体。[化学式31]
[0032]
式(i)所示的化合物亦包含式(i)所示的化合物(亚氨基体)与式(i')所示的化合物(氨基体)的混合物,可以任意比率进行混合。关于式(vi)所示的化合物,亦同样如此。再者,关于式(i)所示的化合物的无水物i型晶体,进行单晶结构分析的结果是确认获得了以下的分子结构(亚氨基体)(关于详情,于实施方案3中记载)。[化学式32]关于式(i)所示的化合物的二水合物晶体及式(i)所示的化合物的无水物ii型晶体,分子结构(氨基体/亚氨基体)未进行鉴定。
[0033]
(粉末x射线衍射(xrpd))一般而言,晶体性有机化合物由在三维空间上周期性地排列的多个分子构成。结构周期性通常表达出能够通过大部分分光学探针(例如,x射线衍射、红外光谱、拉曼光谱及固体nmr)明确地区别的物理特性。其中,粉末x射线衍射(xrpd)为用于测定固体晶体性的敏感度最佳的分析法之一。若x射线照射至晶体上,则于晶格面反射,并相互干涉,仅满足由布拉格定律所预测的条件的方向上的衍射线的强度增大,呈现出与结构周期对应的有序的衍射线。另一方面,对于非晶质固体,未确认到有序的衍射线。非晶质固体通常于其结构之中不具有有序的重复周期,因而不会产生衍射现象,呈现出无特征的较宽的xrpd图案(也称为晕圈图案(halo pattern))。
[0034]
式(i)所示的化合物的无水物的晶体形态可通过粉末x射线衍射图案或特征峰赋予特征。式(i)所示的化合物的无水物的晶体形态因特征性衍射峰的存在而能够与其他晶体形态(例如水合物晶体等)区别。本说明书中所使用的特征性衍射峰为自所观察到的衍射图案中所选择的峰。于区分复数个晶体时,于该晶体中发现但在其他晶体中未发现的峰成为确定该晶体时优选的特征峰,而不是峰的大小。只要为这样的特征峰,即便一个或两个峰亦能够对该晶体赋予特征。对所测得的图进行比较,若所述特征峰一致,则可谓粉末x射线
衍射光谱实质上一致。
[0035]
一般而言,粉末x射线衍射中的衍射角度(2θ)可能会在
±
0.2
°
的范围内产生误差,因此需要理解为粉末x射线衍射的衍射角度的值亦包含
±
0.2
°
左右的范围内的数值。因此,本发明不仅包含粉末x射线衍射中的峰的衍射角度完全一致的晶体,亦包含峰的衍射角度在
±
0.2
°
左右的误差内一致的晶体。
[0036]
已知以下的表及图中所表示的峰的强度通常可能因诸多因素,例如晶体对x射线光束的选择性配向的效果、粗大粒子的影响、供分析的物质的纯度或样品的结晶度而有所变动。另外,关于峰位置,亦可能基于样品的高度变动而位移。进而,若使用不同的波长进行测定,则根据布拉格式(nλ=2dsinθ)获得不同的位移,这样的由于使用不同的波长而获得的不同的xrpd图案亦包含于本发明的范围内。
[0037]
单晶结构分析(参照樱井敏雄著“x射线结构分析指南”裳华房发行(1983年),stout&jensen著x-ray structure determination:a practical guide,macmillan co.,new york(1968)等)为确定晶体的方法之一,可获得该晶体的晶体学参数,进而获得原子坐标(表示各原子的空间位置关系的值)及三维结构模型。在对如本发明的复合体的晶体结构进行鉴定时,可使用单晶结构分析。
[0038]
(拉曼光谱法)拉曼光谱显示分子或复合体系的振动的特征。其起源在于分子与包含光线的光的粒子即光子之间的非弹性碰撞。分子与光子的碰撞带来能量的交换,其结果为能量产生变化,由此光子的波长产生变化。即,拉曼光谱为光子入射至对象分子时所发出的波长极窄的光谱线,因此作为光源,可使用激光等。各拉曼线的波长通过自入射光的波数位移来表示,其为拉曼线与入射光的波长的逆数之间的差。拉曼光谱为测定分子的振动状态者,其由该分子结构来确定。一般而言,拉曼光谱中的吸收带(cm-1
)在
±
2cm-1
的范围内可能会产生误差,因此需要理解为,上述吸收峰的值亦包含
±
2cm-1
左右的范围内的数值。因此,本发明不仅包含拉曼光谱中的吸收带的峰完全一致的晶体,亦包含吸收带的峰在
±
2cm-1
左右的误差内一致的晶体。
[0039]
(红外吸收光谱测定法(ir法))红外吸收光谱测定法是针对各波数来测定红外线通过试样时被吸收的程度的方法。红外吸收光谱通常以横轴上记录波数、纵轴上记录透射率或吸亮度的图表来表示。吸收峰的波数及透射率(或吸亮度)除可在图表上读取以外,还可使用通过数据处理装置而得的算出值。红外吸收光谱由该物质的化学结构来确定。因此,可测定各种波数下的吸收来对物质进行确认或定量。多晶型的判别可通过对该多晶型上特征性官能团,即主要与晶体结构中的氢键相关的官能团例如c=o键、oh键及nh键等,以及其他特征性官能团,例如,c-x(卤素)键、c=c键及c≡c键等的吸收带进行比较来进行。自与特征性官能团对应的约20个吸收峰、更优选为约10个吸收峰、最优选为约5个吸收峰进行选择。通常,试样的吸收光谱在波数4000cm-1
~400cm-1
的范围内进行测定。吸收光谱的测定在与进行装置的分辨率、波数刻度及波数精度的确认时相同的操作条件下进行。
[0040]
一般而言,红外吸收光谱测定中的吸收带(cm-1
)在
±
2cm-1
的范围内可能会产生误差,因此需要理解为,上述吸收峰的值亦包含
±
2cm-1
左右的范围内的数值。因此,本发明不
仅包含红外吸收光谱测定中的吸收带的峰完全一致的晶体,亦包含吸收带的峰在
±
2cm-1
左右的误差内一致的晶体。
[0041]
红外吸收光谱的测定方法有溴化钾片剂法、溶液法、浆料法、液膜法、薄膜法、气体试样测定法、atr法、扩散反射法等。其中,atr法(attenuated total reflection,减弱全反射)称为全反射测定法,为反射法之一。该方法是使试样与由如krs-5的高折射率的物质所制作的棱镜的表面密接,使光以临界角以上的角度入射至棱镜,测定于棱镜与试样的边界产生全反射的光,获得吸收光谱的方法。可利用atr法进行测定的条件之一为棱镜的折射率大于试样,因此需要根据试样来变换棱镜的材质。另外,作为其他条件,棱镜与试样必须密接。因此,适合于液体、粉末、塑料、柔软的橡胶等的测定,具有于不对试样进行化学或物理处理的情况下便可进行测定的优势。另一方面,扩散反射法是在粉末试样的测定中不制作溴化钾片剂,于粉末状态下直接测定的方法。若对试样照射光,则会产生于粉末表面正反射而射向外部的光、及进入试样内部并反复进行透射及扩散后射向表面的扩散反射光(散射光),于扩散反射法中,使用后者获得吸收光谱。
[0042]
(固体
13
c-nmr(核磁共振))固体
13
c-nmr由于(i)光谱数与对象化合物的碳数一致、(ii)化学位移范围宽于1h-nmr、(iii)信号较固体1h-nmr明显,即便包含(iv)添加物,于不相互作用的情形时,化学位移亦不变化等,因此对晶体形式的特定有用。再者,根据所使用的特定的分光计及分析者的试样制备技法,可预测所观察到的化学位移稍有变动。固体
13
c-nmr光谱的误差范围为约
±
0.5ppm。
[0043]
(示差扫描量热测定法(dsc))dsc为热分析的主要测定方法之一,是对作为原子
·
分子的集合体的物质的热性质进行测定的方法。通过dsc测定药物活性成分的与温度或时间相关的热量变化,并将所获得的数据相对于温度或时间进行绘图,由此获得示差扫描量热曲线。根据示差扫描量热曲线可获得关于药物活性成分熔解时的设定温度(on-set temperature)、伴随熔解的吸热峰曲线的最大值及焓的信息。关于dsc,已知观察到的温度可能取决于温度变化速度以及所使用的试样制备技法及特定的装置。因此,dsc中的“熔点”是指不易受试样的制备技法的影响的设定温度。根据示差扫描量热曲线所获得的设定温度的误差范围为约
±
2℃。于晶体的同一性的认定中,不仅熔点,整体的图案亦重要,因测定条件或测定机器而可能多少有所变化。
[0044]
(示差热热重量同步测定法(tg/dta))tg/dta为热分析的主要测定方法之一,是测定作为原子
·
分子的集合体的物质的重量及热性质的方法。tg/dta为测定药物活性成分的与温度或时间相关的重量及热量变化的方法,通过将所获得的数据相对于温度或时间进行绘图,可获得tg(热重量)及dta(示差热)曲线。根据tg/dta曲线,可获得与药物活性成分的分解、脱水、氧化、还原、升华、蒸发相关的重量及热量变化的信息。关于tg/dta,已知所观察到的温度、重量变化可能取决于温度变化速度以及所使用的试样制备技法及特定的装置。因此,tg/dta中的“熔点”是指不易受试样的制备技法的
影响的设定温度。于晶体的同一性的认定中,不仅熔点,整体的图案亦重要,因测定条件或测定机器而可能多少有所变化。
[0045]
(水分吸附脱附等温线测定法(dvs))水分吸附脱附等温线测定是通过针对测定对象的固体测定各相对湿度条件下的重量变化来测量水分的吸附、脱附行为的测定法。作为基本测定法,可以0%rh(相对湿度0%)下的干燥重量为基准,每5%或10%地提高相对湿度,于各相对湿度下的重量稳定化后,根据自基准值的重量增加求出吸附水的量。同样,可通过自100%rh每5%或10%地降低相对湿度,来测定水的脱附量。通过对各相对湿度下的重量变化值进行绘图,能够获得吸附脱附等温线。根据该结果,能够探讨各湿度下的附着水分的吸附、脱附现象。另外,于无水物晶体因湿度而与水合物晶体相互进行晶体转变的情形时,能够计算出产生晶体转变的湿度、及结晶水的量。由于附着水、及结晶水的吸附脱附受粒径、结晶度、晶体习性等的影响,因此测定结果可能多少有所变化。
[0046]
含有本发明的晶体的药物组合物作为慢性咳嗽的治疗剂或预防剂非常有用。
[0047]
本发明的晶体可以其本身对人类患者进行投予、或可以将该晶体与适当的载体或赋形剂混合而成的药物组合物进行投予。药物的配方及用于投予的技术可将本领域技术人员所知的制剂配方或技术加以组合并适当选择来使用。
[0048]
本发明的晶体或含有它们的药物组合物的投予路径并不限定,可包含经口、直肠、经黏膜或肠投予或者肌内、皮下、脊髓内、鞘内、直接性心室内、静脉内、玻璃体内、腹腔内、鼻腔内、眼内、及注射。优选的投予路径为经口。
[0049]
本发明的药物组合物可通过本领域中广为人知的制法,例如惯用的混合、溶解、颗粒化、糖衣-制作、粉末化、乳化、胶囊化、覆埋、冷冻干燥工艺来制造。
[0050]
本发明的晶体或含有它们的药物组合物可使用水性溶液,优选为生理学上适合的林格氏溶液或生理盐水这样的缓冲液,通过注射进行投予。
[0051]
本发明的晶体或含有它们的药物组合物可使用与应渗透的屏障适合的渗透剂进行经黏膜投予。该渗透剂通常可使用本领域中已知者。
[0052]
本发明的晶体或含有它们的药物组合物可通过配合本领域中广为人知的药物上可接受的载体进行经口投予。可根据该载体,将本发明的晶体制成片剂、丸剂、口含片、糖衣片、胶囊剂、液剂、凝胶、糖浆、悬浮剂进行投予。经口投予用的药物组合物可通过如下方式来制作:使用固体赋形剂,若有需要,添加其他适当的辅助剂,其后,将所获得的混合物进行粉碎,并对颗粒的混合物进行处理获得片剂或糖衣片核。
[0053]
有用的赋形剂特别是包含乳糖、蔗糖、甘露糖醇或山梨糖醇的糖等填充剂,例如为玉米淀粉、小麦淀粉、米淀粉及马铃薯淀粉等纤维素制备物,明胶、黄蓍胶、甲基纤维素、羟丙基甲基纤维素和/或羧甲基纤维素钠等。若有需要,可添加琼脂、海藻酸等崩解剂。亦可使用海藻酸钠等盐。
[0054]
可用于经口投予的药物组合物包括用明胶所制作的推入配合式胶囊(push-fit capsule)、用明胶及甘油或山梨糖醇等塑化剂所制作的密封胶囊等。推入配合式胶囊可包含与乳糖等填充剂、淀粉等黏合剂和/或滑石或硬脂酸镁等润滑剂混合而成的有效成分。
[0055]
药物组合物亦可包含适当的固体或凝胶相的载体或赋形剂。这样的载体或赋形剂
例如可例举:碳酸钙、磷酸钙、各种糖、淀粉、纤维素衍生物、明胶、聚乙二醇等聚合物。
[0056]
在本发明的晶体或其药物组合物中,治疗有效量最初可根据细胞培养分析进行估计。继而,可以达成包括在细胞培养中所确定的ic50(即,本发明的晶体或其药物组合物达成抑制pk活性的最大值一半的浓度)的循环浓度范围的方式加大投予量,以在动物模型中使用。继而,可使用这样的信息更准确地确定人类的有用剂量。
[0057]
本发明的晶体或其药物组合物的治疗效果可在细胞培养或实验动物中通过标准药物方法进行测定。例如,可根据专利文献9中记载的生物试验方法进行评价。自所述细胞培养分析及动物实验所获得的资料可用于规定用于人类的投予量范围。投予量可根据所使用的投予形态及所利用的投予路径而变化。关于准确的配方投予路径及投予量,可由各医生考虑患者的状态而进行选择。
[0058]
可将本发明的晶体或其药物组合物与其他药剂加以组合以治疗疾病及障碍亦为本发明的方面。
[0059]
根据本发明,可提供式(i)所示的化合物的无水物晶体或水合物晶体。该晶体性固体至少具有以下的任一特征。(1)对热、湿度、溶剂、光等的稳定性良好,保存稳定性较高。(2)着色稳定性良好。(3)对水或有机溶剂的溶解度良好。(4)对水或有机溶剂的溶解速度较快。(5)为高纯度。(6)有机溶剂的残存率较低。(7)过滤、离心分离、制剂化等的操作性优异。(8)比容较小。(9)不易带电。(10)可在环境负荷较少的条件下以高产率制造,可大量制造。(11)作为注射剂等的药物活性成分或其制造用原体有用。(12)由于能够控制至不伴随血管痛的静脉注射所适合的ph值范围,因此对进行制剂时的液量控制或赋形剂的削减等有利。(13)流动性良好。(14)压缩度(%)较低。特别是本发明的晶体性固体即便于较宽的湿度范围(例:25~99%rh等)或过于苛刻的环境下(例:多湿下)亦稳定性较高。
[0060]
以下对本说明书中所使用的各用语的含义进行说明。各用语只要未特别申明,则单独使用的情形、或与其他用语组合使用的情形均以相同的含义来使用。用语“由
……
所组成”意指仅具有构成要件。用语“包含”意指不限定于构成要件,不排除未记载的要素。
[0061]
以下,针对本发明一面公开实施方案一面进行说明。涵盖整个本说明书,关于单数形式的表达,只要未特别提及,则应理解为亦包含其复数形式的概念。因此,关于单数形式的冠词(例如,于英语的情形时,“a”、“an”、“the”等),只要未特别提及,则应理解为亦包含其复数形式的概念。
另外,关于本说明书中所使用的用语,只要未特别提及,则应理解为以上述本领域中通常所使用的含义来使用。因此,只要未另外进行定义,本说明书中所使用的所有专业用语及科学技术用语具有与由本领域技术人员通常所理解的含义相同的含义。在矛盾的情形时,本说明书(包括定义)优先。
[0062]“卤素”包含氟原子、氯原子、溴原子、及碘原子。特别优选为氟原子及氯原子。
[0063]“烷基”包含碳数1~15优选为碳数1~10、更优选为碳数1~6、进一步优选为碳数1~4的直链或支链状的烃基。例如可例举:甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、正己基、异己基、正庚基、异庚基、正辛基、异辛基、正壬基、正癸基等。作为“烷基”的优选的方面,可例举:甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基。作为进而优选的方面,可例举:甲基、乙基、正丙基、异丙基、叔丁基。“c1-c4烷基”可例举:甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基或叔丁基。
[0064]
本发明包括制造式(iv):[化学式35](式中,r1为c1-c4烷基)所示的化合物或其盐的步骤,该步骤的特征在于:使式(ii):[化学式33]所表示的化合物或其盐与式(iii):[化学式34](式中,r1为c1-c4烷基)所示的化合物或其盐,在选自由氯化锂、氯化钙、氯化镁、溴化锂、对甲苯磺酸、甲磺酸及三氟甲磺酸所组成的组的1种以上添加剂的存在下反应。式(ii)所示的化合物或其盐、及式(iii)所示的化合物或其盐可利用市售试剂并按照公知的方法来制造、或可使用市售品。
[0065]
作为溶剂,只要不阻碍反应,则无特别限定,可使用甲醇、乙醇、异丙醇、叔丁醇、及它们的混合溶剂。例如可使用甲醇。
[0066]
反应温度通常是在室温至溶剂回流的温度的范围内实施。例如可在-10℃~溶剂回流的温度的范围内进行。例如可在80℃进行。
[0067]
反应时间为1~20小时,例如为5~7小时。
[0068]
相对于式(ii)所示的化合物,式(iii)所示的化合物的使用量通常可使用1.0~10.0当量,例如2.0~4.0当量,例如3.0当量。
[0069]
作为添加剂,可使用氯化锂、氯化钙、氯化镁、溴化锂、对甲苯磺酸、甲磺酸、三氟甲磺酸等。可选择复数种所述添加剂同时使用。相对于式(ii)所示的化合物,添加剂的使用量通常可使用0.1~5.0当量,例如1.0~2.0当量,例如1.0~1.5当量。
[0070]
本发明包括制造式(iv-a):[化学式37](式中,r1为c1-c4烷基)所示的化合物的对甲苯磺酸盐的步骤,该步骤的特征在于:在式(iv):[化学式36](式中,r1为c1-c4烷基)所示的化合物或其盐中添加对甲苯磺酸。相对于式(ii)所示的化合物,对甲苯磺酸一水合物(或对甲苯磺酸水溶液)的使用量通常可使用0.5~2.0当量,例如0.8~1.0当量。
[0071]
本发明包括制造式(v):[化学式39]所表示的化合物的1/2硫酸盐的步骤,该步骤的特征在于:对式(iv-b):[化学式38]所表示的化合物的对甲苯磺酸盐进行氢化分解反应,并添加硫酸。式(iv-b)所示的化合物可按照上述步骤制造。
[0072]
作为溶剂,只要不阻碍反应,则无特别限定,可使用甲醇、乙醇、1-丙醇、异丙醇、叔丁醇、四氢呋喃及它们的混合溶剂。例如,可使用甲醇。
[0073]
反应温度通常是在室温至溶剂回流的温度的范围内实施。例如可在室温~溶剂回流的温度的范围内进行。例如可在30~50℃下进行。
[0074]
反应时间为30分钟~20小时,例如为1~3小时。
[0075]
作为氢解反应触媒,可使用钯碳、氢氧化钯、钯黑等。相对于式(iv-b)所示的化合物,氢解反应触媒的使用量通常可使用0.01~1w/w,例如0.1~0.3w/w。
[0076]
相对于式(iv-b)所示的化合物,浓硫酸的使用量通常可使用0.01~0.5当量,例如0.3~0.4当量。
[0077]
本发明包括制造式(i):[化学式41]所表示的化合物或其盐的步骤,该步骤的特征在于:使式(vi):[化学式40](式中,r1为c1-c4烷基)所示的化合物或其盐,在选自由异丙醇、四氢呋喃及叔丁醇所组成的组的1种以上溶剂的存在下进行水解反应。式(vi)所示的化合物可按照上述步骤及专利文献6、7、8及9中记载的方法制造。
[0078]
作为溶剂,只要不阻碍反应,则无特别限定,可使用异丙醇(2-丙醇)、四氢呋喃及叔丁醇及它们的混合溶剂。例如可使用异丙醇(2-丙醇)。
[0079]
反应温度通常可在-10℃至溶剂回流的温度下进行。例如可在30℃~40℃下进行。
[0080]
反应时间为0.1~20小时,例如为1~5小时。
[0081]
作为碱,可使用氢氧化钠、氢氧化钾、氢氧化锂等。例如可使用氢氧化钠。相对于式(vi)所示的化合物,碱的使用量通常可使用2.0~5.0当量,例如2.0~3.0当量。实施例
[0082]
通过以下的实施方案进一步详细地说明本发明。所述并不对本发明进行限定。关于数值(例如,量、温度等),应考虑到些许误差及偏差。只要未特别说明,则%为成分的重量%及组合物的总重量的重量%,压力为大气压或与其相近的压力。
[0083]
(粉末x射线衍射图案的测定)依照日本药典的一般试验法中所记载的粉末x射线衍射测定法对各实施方案中所获得的晶体进行粉末x射线衍射测定。将测定条件示于以下。(方法1)(装置)rigaku公司制造的smartlab(操作方法)测定法:反射法使用波长:cukα射线管电流:200ma管电压:45kv试样板:玻璃x射线的入射角:2.5
°
取样间隔:0.02
°
检测器:hypix-3000(二维检测模式)(方法2)(装置)bruker公司制造的d-8discover(操作方法)测定法:反射法使用波长:cukα射线管电流:40ma管电压:40kv试样板:铝x射线的入射角:3
°
及12
°
(方法3)bruker公司制造的d-8discover(操作方法)测定法:反射法使用波长:cukα射线管电流:40ma管电压:40kv试样板:铝x射线的入射角:3
°
[0084]
(拉曼光谱的测定)进行各实施例中所获得的晶体的拉曼光谱的测定。将测定条件示于以下。(方法1)测定机器:labram aramis(horiba jobin yvon公司制造)测定方法:显微激光拉曼光谱法
激光波长:633nm(he-ne激光)衍射光栅:600grooves/mm检测器:ccd检测器物镜:20
×
(na 0.25)累计次数:5次曝光时间:5秒(方法2)测定机器:ramantouch vis2-nir-snu(nanophoton公司制造)测定方法:显微激光拉曼光谱法激光波长:532nm衍射光栅:1200grooves/mm检测器:ccd检测器物镜:20
×
(na0.45)累计次数:1次曝光时间:3秒
[0085]
(示差扫描量热(dsc)的测定)进行各实施例中所获得的晶体的dsc的测定。称取试样约4.199mg至铝锅中,简易密封以进行测定。将测定条件示于以下。需要说明的是,通过示差扫描量热(dsc)的测定有可能在
±
2℃的范围内会产生误差。装置:ta instruments discovery测定温度范围:0℃-220℃升温速度:10℃/分钟氛围:n
2 50ml/分钟
[0086]
(nmr测定)存在公开nmr数据的情形时未记载所测得的所有的峰的情况。
[0087]
(hplc测定)(方法a)柱:xbridge c18,φ4.6
×
150mm,3.5μm(waters)柱烘箱:40℃流量:每分钟1.0mluv检测波长:254nm流动相a:0.1%三氟乙酸水溶液流动相b:液相色谱用乙腈将梯度程序示于表1。
[0088]
[表1]注入后的时间(分)流动相a(vol%)流动相b(vol%)0~485154~1085
→
6015
→
4010~1360
→
1040
→
90
13~17109017~17.0110
→
8590
→
1517.01~278515
[0089]
(方法b)柱:chiralpack as-rh,φ4.6
×
150mm,5μm(daicel化学)柱烘箱:35℃流量:每分钟1.0mluv检测波长:254nm流动相a:液相色谱法用纯化水流动相b:液相色谱用乙腈将梯度程序示于表2。
[0090]
[表2]注入后的时间(分钟)流动相a(vol%)流动相b(vo1%)0~14802014~1880
→
1020
→
9018~24109024~24.0110
→
8090
→
2024.01~308020
[0091]
(方法c)柱:chiralpack ic,φ4.6
×
250mm,5μm(daicel化学)柱烘箱:35℃流量:每分钟1.0mluv检测波长:262nm流动相:0.1%甲酸水溶液/液相色谱用乙腈混合液(3:2)
[0092]
(方法d)柱:cadenza cd-c18,φ3.0
×
150mm,3μm柱烘箱:50℃流量:每分钟0.55mluv检测波长:262nm流动相a:0.1%tfa水溶液流动相b:乙腈将梯度程序示于表3。
[0093]
[表3]时间(分钟)流动相a(%)流动相b(%)0802018020104555154555201090
25109025.018020308020需要说明的是,需要理解,hplc的保留时间包含些许误差。
[0094]
(tg/dta的测定)称取实施例7中所获得的晶体约4.4mg,装入至铝锅,于开放系统中进行测定。测定条件如下所示。装置:hitachi high-technologies tg/dta sta7200rv测定温度范围:室温-300℃升温速度:10℃/分钟
[0095]
(单晶结构分析的测定及分析方法)将单晶结构分析的测定条件及分析方法如下所示。(装置)rigaku公司制造xtalab p200 mm007(测定条件)测定温度:25℃使用波长:cukα射线软件:crysalispro 1.171.39.46e(rigaku oxford diffraction,2018)(数据处理)软件:crysalispro 1.171.39.46e(rigaku oxford diffraction,2018)数据进行过劳伦兹及偏光修正、吸收修正。(晶体结构分析)使用直接法程序shelxt(sheldrick,g.m.,2015)进行相位确定,精密化为使用shelxl(sheldrick,g.m.,2015)实施全矩阵(full-matrix)最小平方法。非氢原子的温度因子均以各向异性进行精密化。氧o5上的氢原子h5为自差傅立叶图导出,并进行了精密化。其余氢原子为使用shelxl的预设参数通过计算进行导入,将其视为骑乘原子(riding atom)进行处理。所有氢原子为以各向同性参数进行精密化。r1(i>2.00s(i))为0.0470,根据最终的差傅立叶,确认并无电子密度的缺少或错位。图7及图8的制图中使用了pluton(spek,1991)/ortep(johnson,1976)(50%机率水平)。实施例1
[0096]
化合物(3)的合成[化学式42][1]化合物(3)的合成
在室温下,于(r)-(+)-1-苯基乙基胺(1)(20.01g,165.1mmol)中加入甲醇(20ml)、及甲基丙烯酸甲酯(2)(49.64g,495.8mmol)。冷却至-10℃后加入氯化锂(7.07g,167mmol)。将反应液升温至80℃,并搅拌6小时。将反应液冷却至25℃,加入9.1%氯化钠水溶液(77.03g),通过分液将水层去除。在室温下,于所获得的水层中加入甲苯(61.04g),并通过分液将水层去除。将所获得的2个有机层合并,加入甲苯(16.99g)并于50℃下进行减压蒸馏去除。将对甲苯磺酸一水合物(28.91g,152.0mmol)溶解于乙醇(16.00g)中,制备对甲苯磺酸的乙醇溶液(44.91g)。在上述所制备的浓缩液中加入甲苯(155.91g),在室温加入在上述所制备的对甲苯磺酸的乙醇溶液(5.88g)及化合物(3)的种晶(19.95mg,0.05070mmol)中加入甲苯(63μl)使其悬浮而得的浆料。于所获得的浆料中加入上述所制备的对甲苯磺酸的乙醇溶液(39.93g),加入乙醇(10ml)并搅拌2小时,彻夜放置。冷却至0℃并搅拌2小时,滤取固体,以粗产物的形式获得化合物(3)(20.75g,31.9%)。在化合物(3)的粗产物的一部分(5.00g)中加入甲苯(1.92g)、乙酸乙酯(21.70g)及甲醇(2.90g),于50℃下搅拌3小时。冷却至0℃,滤取固体而获得化合物(3)(4.65g)。元素分析:c 61.22%,h 7.09%,n 3.56%,s 8.12%1h-nmr(dmso-d6)δppm:1.12(d,j=7.0hz,3h),1.55(br d,j=6.7hz,3h),2.29(s,3h),2.50(s,2h),2.83(br dd,j=13.2hz,6.9hz,1h),2.90(m,2h),3.62(s,3h),7.13(m,2h),7.47(m,7h)
[0097]
[2]化合物(3)的种晶的合成将(r)-(+)-1-苯基乙基胺(1)(2.00g,16.5mmol)、甲醇(1.59g)、甲基丙烯酸甲酯(2)(4.97g,49.6mmol)及氯化锂(0.70g,17mmol)在室温下加以混合,升温至80℃并搅拌4小时。将反应液冷却至25℃,加入9.1%氯化钠水溶液(7.70g),通过分液将水层去除。于所获得的有机层中加入甲苯(5.21g)后,加入将对甲苯磺酸一水合物(2.88g,15.1mmol)溶解于甲醇(1.58g)中所调整的对甲苯磺酸的甲醇溶液(4.46g)。将该反应液加入至冷却至0℃的甲苯(6.94g)中,于0℃下搅拌30分钟。滤取所析出的固体,由此获得化合物(3)的种晶(1.60g,24.6%)。[实施例1-1]
[0098]
化合物(3)的合成[化学式43]在室温下,在(r)-(+)-1-苯基乙基胺(1)(20.00g,165.0mmol)中加入对甲苯磺酸一水合物(1.57g,8.25mmol)、及甲基丙烯酸甲酯(2、49.57g,495.1mmol),升温至99℃,并于99℃下反应16小时。冷却至25℃后,于反应液中加入甲醇(8ml),其后与将对甲苯磺酸一水合物(27.31g,143.6mmol)溶解于乙酸乙酯(40ml)及甲醇(4ml)中而得的对甲苯磺酸溶液进
行混合。于25℃下,于该反应液中加入乙酸乙酯(100ml),滤取所析出的固体,以粗产物的形式获得化合物(3)(28.14g)。于化合物(3)的粗产物(28.14g)中加入乙酸乙酯(15ml)及甲醇(30ml),升温至60℃之后冷却至40℃,于40℃下加入将乙酸乙酯(60μl)加入至化合物(3)的种晶(20.0mg,0.308mmol)中使其悬浮而得的浆料。于40℃下对所获得的浆料搅拌30分钟,其后冷却至22℃,加入乙酸乙酯(218ml),冷却至0℃并搅拌1小时。滤取所析出的固体,由此获得化合物(3)(22.29g,34.32%)。实施例2
[0099]
化合物(4)的合成[化学式44]在化合物(3)(22.00g,55.91mmol)中加入甲苯(95.26g)及水(44.00g)使其悬浮,加入8%氢氧化钠水溶液(27.54g)及水(4.40g),并通过分液将水层去除。于所获得的有机层中加入水(11.00g),并通过分液将水层去除。将所获得的有机层以50℃进行减压蒸馏去除,重复进行加入甲醇的操作,溶剂置换为甲醇。于所获得的浓缩液中加入浓硫酸(2.04g,19.8mmol)、10%钯碳(2.20g,约40%湿润品)及甲醇(17.41g)。将反应液升温至40℃,于氢气氛围下搅拌90分钟。通过过滤将钯碳去除,并于所获得的滤液中加入甲醇(52.24g)及浓硫酸(0.67g,6.5mmol)。于所获得的反应液中加入乙腈,重复进行减压蒸馏去除的操作,溶剂置换为乙腈,并冷却至0℃。滤取所析出的固体,由此获得化合物(4)(8.58g,92.3%)。元素分析:c 35.72%,h 7.18%,n 8.55%,s 9.63%1h-nmr(dmso-d6)δppm:1.10(d,j=7.1hz,3h),2.62(m,1h),2.75(dd,j=12.7h,5.9hz,1h),2.89(dd,j=12.7hz,7.3hz,1h),3.63(s,3h)
[0100]
(参考例1)化合物(5)的合成[化学式45]使化合物(4)(19.00g,114.3mmol)悬浮于乙腈(45.00g)中。以2℃加入1,8-二氮杂双环[5.4.0]-7-十一烯(19.10g,125.5mmol)及乙腈(3.00g),于2℃下搅拌30分钟。将反应液以2℃加入至使n,n-羰基二咪唑(21.30g,131.4mmol)悬浮于乙腈(75.00g)中而得的浆料中。于反应液中加入乙腈(15.00g),于2℃下搅拌1小时22分钟。于反应液中以2℃加入1,8-二氮杂双环[5.4.0]-7-十一烯(17.40g,114.3mmol)及乙腈(3.00g),并冷却至1℃。于反应液中加入1h-吡唑-1-羧基脒盐酸盐(16.80g,114.6mmol)及乙腈(3.00g)。将反应液升温至
60℃,搅拌2小时10分钟。将反应液冷却至20℃。于反应液中以2℃加入1,8-二氮杂双环[5.4.0]-7-十一烯(27.80g,182.6mmol)及乙腈(3.00g),并冷却至-10℃。于反应液中加入n,n-羰基二咪唑(29.70g,183.2mmol)及乙腈(3.00g)。将反应液于2℃下搅拌1小时20分钟。于反应液中以2℃加入甲醇(7.50g)、乙酸(4.80g,79.9mmol)及乙腈(3.00g)。将反应液以50℃进行减压蒸馏去除。于所获得的浓缩液中加入n,n-二甲基乙酰胺(27.00g),冷却至10℃并加入17%硫酸水(204.1g)及水(19.00g)。于反应液中以25℃加入17%硫酸水(31.30g)及水(2.50g),并搅拌1小时48分钟。将反应液以50℃进行减压蒸馏去除。于所获得的浓缩液中加入水(190ml)并冷却至2℃后,加入17%硫酸水(3.30g)及水(1.30g)。将反应液于2℃下搅拌1小时15分钟,滤取所析出的固体而获得化合物(5)(27.13g,85.0%)。1h-nmr(cdcl3)δppm:1.24(d,j=7.1hz,3h),3.02(m,1h),3.68(s,3h),4.02(dd,j=13.4hz,6.3hz,1h),4.24(dd,j=13.3hz,8.4hz,1h),6.60(dd,j=2.8hz,1.6hz,1h),7.85(d,j=1.6hz,1h),8.48(dd,j=2.9hz,0.6hz,1h),9.70(brs,1h)
[0101]
(参考例2)化合物(8)的合成[化学式46][1]化合物(8)的合成使叔丁醇钠(12.50g,130.1mol)悬浮于n-甲基-2-吡咯烷酮(64.00g)中,并加入4-氨基苯酚(7)(14.10g,129.2mmol)及n-甲基-2-吡咯烷酮(16.00g)。将反应液升温至100℃,并加入2-溴吡啶(6)(19.50g,123.4mmol)及n-甲基-2-吡咯烷酮(4.00g)。将反应液于115℃下搅拌8小时20分钟,并冷却至50℃。于反应液中以50℃加入水(29.00g),冷却至25℃并加入水(107.00g)。于反应液中加入化合物(8)的种晶(8,20mg)及水(195mg),于20℃下搅拌50分钟。于反应液中以25℃加入水(156.00g),冷却至5℃并搅拌1小时30分钟。滤取所析出的固体,由此获得化合物(8)(17.81g,77.5%)。1h-nmr(cdcl3)δppm:3.60(s,2h),6.69-6.73(m,2h),6.83(ddd,j=8.4hz,0.8hz,0.8hz,1h),6.92-6.96(m,3h),7.63(ddd,j=8.0hz,7.2hz,2.0hz,1h),8.18(ddd,j=5.2hz,2.0hz,0.8hz,1h)
[0102]
[2]化合物(8)的种晶合成使叔丁醇钠(3.20g,33.3mmol)悬浮于n-甲基-2-吡咯烷酮(16.42g)中,并加入4-氨基苯酚(7)(3.64g,33.4mmol)及n-甲基-2-吡咯烷酮(4.12g)。将反应液升温至100℃,并加入2-溴吡啶(6)(5.01g,31.7mmol)及n-甲基-2-吡咯烷酮(1.10g)。将反应液于115℃下搅拌6小时,加入叔丁醇钠(1.07g,11.1mmol)于115℃下搅拌2小时35分钟后冷却至50℃。于反应液中以50℃加入水(7.50g)后冷却至25℃,加入水(67.54g),冷却至1℃使其晶体化。将所获得的浆料于5℃下搅拌30分钟后,滤取所析出的固体,由此获得化合物(8)的种晶(3.84g,65.2%)。
[0103]
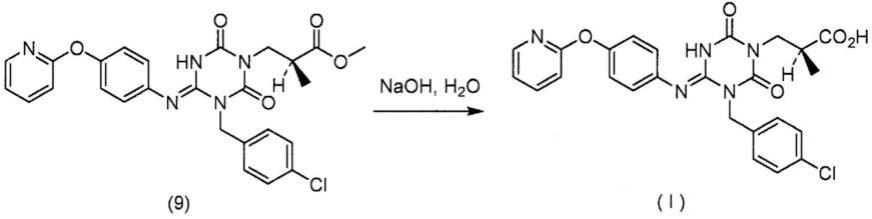
(参考例3)化合物(9)的合成[化学式47]
[1]化合物(9)的合成在化合物(5)(10.13g,36.27mmol)中加入溴化钠(4.1g,39.9mmol)及n,n-二甲基乙酰胺(27.70g)。在反应液中加入n,n-二异丙基乙基胺(5.16g,39.9mmol)及n,n-二甲基乙酰胺(0.96g),并升温至75℃。在反应液中于75℃下加入将4-氯苄氯(6.43g,39.9mmol)溶解于n,n-二甲基乙酰胺(9.56g)中所制备的4-氯苄氯的n,n-二甲基乙酰胺溶液,并加入n,n-二甲基乙酰胺(9.56g)。将反应液于75℃下搅拌5小时15分钟。将反应液冷却至25℃,加入乙酸(0.65g,11mmol)并升温至40℃。于反应液中加入将n,n-二甲基乙酰胺(9.55g)溶解于化合物(8)(7.43g,39.9mmol)中所制备的化合物(8)的n,n-二甲基乙酰胺溶液,并加入n,n-二甲基乙酰胺(9.55g)。将反应液于40℃下搅拌3小时,并冷却至室温。于反应液中加入丙酮(27.94g)及水(35.46g)。于反应液中加入化合物(9)的种晶(10.13mg)、水(0.40g)及丙酮(0.08g),在室温下搅拌3小时25分钟后,彻夜放置。将反应液在室温下搅拌1小时后,加入水(30.39g)搅拌3小时25分钟。滤取所析出的固体而获得化合物(9)(16.72g,88.3%)。1h-nmr(cdcl3)δppm:1.19(d,j=7.1hz,3h),2.91(m,1h),3.61(s,3h),3.90(dd,j=13.6hz,6.2hz,1h),4.12(dd,j=13.6hz,8.4hz,1h),5.18(d,j=14.2hz,1h),5.22(d,j=14.2hz,1h),6.85(m,2h),6.96(m,1h),7.00(m,1h),7.14(m,2h),7.31(m,2h),7.50(m,2h),7.70(m,1h),7.89(brs,1h),8.14(m,1h)
[0104]
[2]化合物(9)的种晶合成在化合物(5)(5.01g,17.9mmol)中加入溴化钠(2.00g,19.4mmol)及n,n-二甲基乙酰胺(13.67g)。于反应液中加入n,n-二异丙基乙基胺(2.55g,19.7mmol)及n,n-二甲基乙酰胺(0.47g),并升温至75℃。于反应液中于75℃下加入将4-氯苄氯(3.16g,19.6mmol)溶解于n,n-二甲基乙酰胺(4.71g)中所制备的4-氯苄氯的n,n-二甲基乙酰胺溶液,并加入n,n-二甲基乙酰胺(4.71g)。将反应液于75℃下搅拌4小时30分钟。将反应液冷却至25℃,加入乙酸(0.32g,5.3mmol),并升温至40℃。于反应液中加入将化合物(8)(3.66g,19.7mmol)溶解于n,n-二甲基乙酰胺(4.71g)中所制备的化合物(8)的n,n-二甲基乙酰胺溶液,并加入n,n-二甲基乙酰胺(4.71g)。将反应液于40℃下搅拌3小时25分,并冷却至室温。于反应液中加入丙酮(13.79g)及水(17.54g),在室温下彻夜放置。将反应液升温至25℃并搅拌5小时,加入水(15.00g)于25℃下搅拌2小时。滤取所析出的固体而获得化合物(9)的种晶(8.17g,87.2%)。实施例3
[0105]
式(i)所示的化合物的合成[化学式48]
[1]式(i)所示的化合物的合成于化合物(9)(70.00g,134.1mmol)中加入2-丙醇(109.91g)、水(63.00g)及48%氢氧化钠水溶液(27.94g,335.3mmol)。将反应液升温至35℃,并搅拌4小时10分钟。于反应液中加入2-丙醇(32.97g)、甲醇(177.30g)及水(63.00g)并升温至50℃。于反应液中加入甲酸(18.52g,402.3mmol)及式(i)所示的化合物的种晶(70.00mg),于50℃下搅拌1小时10分钟后加入水(280.00g)并冷却至25℃。滤取所析出的固体而获得式(i)所示的化合物的无水物i型晶体(62.86g,92.3%)。1h-nmr(cdcl3)δppm:1.13(d,j=7.0hz,3h),2.76(m,1h),3.83(dd,j=13.5hz,6.1hz,1h),4.03(dd,j=13.5hz,8.5hz,1h),5.14(m,1h),5.25(d,j=14.4hz,1h),6.82(d,j=8.6hz,2h),7.00(m,2h),7.08(m,2h),7.25(m,2h),7.43(d,j=8.3hz,2h),7.72(m,1h),8.06(dd,j=5.4hz,1.8hz,1h),8.67(brs,1h)
[0106]
[2]式(i)所示的化合物的种晶合成于化合物(9)(1.50g,2.87mmol)中加入甲醇(5.95g)、水(3.00g)及48%氢氧化钠水溶液(0.60g,7.20mmol)。将反应液升温至40℃,并搅拌1小时30分钟。将反应液冷却至室温,在室温下加入甲酸(0.40g,8.62mmol)、乙酸乙酯(10.5ml)、水(9ml),并通过分液将水层去除。于所获得的有机层中加入水(3ml),通过分液将水层去除,于有机层中加入2-丙醇(90ml),并以40℃进行减压蒸馏去除。于所获得的浓缩残渣中加入水(7.5ml)、及2-丙醇(7.5ml)于25℃下搅拌1小时30分钟。加入水(7.5ml)、及甲醇(7.5ml)后,升温至60℃搅拌2小时,冷却至25℃并滤取所析出的固体而获得式(i)所示的化合物的无水物i型晶体的种晶(10,1.25g,85.6%)。
[0107]
将式(i)所示的化合物的无水物i型晶体的粉末x射线衍射的结果示于图1(方法1)。于粉末x射线衍射光谱中,于衍射角度(2θ):12.6
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
、26.6
°±
0.2
°
、27.8
°±
0.2
°
及32.8
°±
0.2
°
、或衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
、17.2
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
、25.4
°±
0.2
°
及27.8
°±
0.2
°
处确认到峰。于上述粉末x射线衍射光谱中,于衍射角度(2θ):15.8
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、23.9
°±
0.2
°
及25.4
°±
0.2
°
、或衍射角度(2θ):7.9
°±
0.2
°
、9.3
°±
0.2
°
、12.9
°±
0.2
°
、15.8
°±
0.2
°
及19.4
°±
0.2
°
处的峰作为式(i)所示的化合物的无水物i型晶体尤其具有特征性。
[0108]
将式(i)所示的化合物的无水物i型晶体的拉曼光谱的结果示于图2(方法1)。于829cm-1
±
2cm-1
、989cm-1
±
2cm-1
、1013cm-1
±
2cm-1
、1093cm-1
±
2cm-1
、1128cm-1
±
2cm-1
、1243cm-1
±
2cm-1
、1370cm-1
±
2cm-1
、1599cm-1
±
2cm-1
、1659cm-1
±
2cm-1
、1735cm-1
±
2cm-1
、2938cm-1
±
2cm-1
、3067cm-1
±
2cm-1
处确认到主要吸收峰。
[0109]
在一个实施方案中,式(i)所示的化合物的无水物i型晶体于829cm-1
±
2cm-1
、989cm-1
±
2cm-1
、1013cm-1
±
2cm-1
、1128cm-1
±
2cm-1
及1370cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的无水物i型晶体于829cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的无水物i型晶体于989cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的无水物i型晶体于1013cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的无水物i型晶体于1128cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的无水物i型晶体于1370cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的无水物i型晶体具有选自由829cm-1
±
2cm-1
的吸收峰、989cm-1
±
2cm-1
的吸收峰、1013cm-1
±
2cm-1
的吸收峰、1128cm-1
±
2cm-1
的吸收峰及1370cm-1
±
2cm-1
的吸收峰所组成的组的1个以上吸收峰。
[0110]
将式(i)所示的化合物的无水物i型晶体的dsc分析结果示于图3。设定温度显示约196℃。
[0111]
将式(i)所示的化合物的无水物i型晶体的单晶结构分析的结果示于以下。将晶体学资料示于表4。[表4]此处,v意指单位晶格体积,z意指单位晶格中的分子数量。
[0112]
另外,将非氢原子的原子座标示于表5~表7。此处,u(eq)意指等价各向同性温度
因子。[表5]原子xyzu(eq)cl22485(2)7006(2)-759.2(19)104.9(7)cl17426.0(16)2947.5(18)10766.8(15)88.8(5)n52089(4)2884(4)7834(3)46.5(9)n97630(4)7464(4)4017(4)50.4(9)n21056(4)4687(4)8118(4)51.9(10)c163580(5)2450(5)9435(4)44.5(10)o55867(3)1451(3)5338(3)63.8(8)n88842(4)5752(4)3669(4)50.7(10)c9394(5)5906(5)7725(5)48.6(11)o21151(4)4145(4)4553(3)65.3(10)o611217(6)534(5)3496(4)86.5(15)c121387(5)4061(5)7442(4)44.9(10)c214123(5)1390(5)10288(4)48.7(11)o33152(4)1150(4)7525(4)69.7(11)o78862(4)6055(4)5375(4)73.6(12)o86672(5)8973(4)2517(4)76.4(12)n31081(4)4424(4)6293(3)47.9(9)c6-895(6)8306(6)7034(5)61.0(14)o45488(6)3422(5)5303(6)122(2)o1-1611(5)9501(4)6620(4)72.8(12)n42263(4)2677(4)5979(3)46.6(9)n1-2337(5)11398(4)6748(4)61.2(11)c11-1514(6)7271(6)7984(5)62.9(14)c397334(5)7962(5)2859(5)53.8(12)c2611309(6)-350(6)2949(6)61.7(14)n107785(4)7278(4)2156(4)54(1)c131468(4)3771(5)5546(4)46.8(11)c2710625(6)-195(6)2050(5)62.2(14)n78777(5)5455(4)1869(4)56.6(11)c195944(5)2756(5)10236(5)56.2(12)c152220(5)2247(5)9106(4)50.8(12)c358767(6)3165(6)3297(5)59.7(13)c205306(6)1541(5)10695(5)58.4(14)c434464(7)6225(6)638(6)77.4(18)c10-861(6)6078(6)8345(5)60.8(14)
[0113]
[表6]
原子xyzu(eq)c142559(5)2162(5)7135(4)47.9(11)c3-797(7)11219(6)8455(6)71.5(16)c425646(7)6422(6)1043(6)68.7(16)c255201(4)2540(5)5041(5)59.2(11)c3310562(6)3952(6)1643(5)63.7(14)c454543(6)8455(6)-693(5)61.4(14)c369349(7)1932(6)3659(6)67.0(14)c378488(5)6084(5)2556(4)48.2(11)c222781(6)1991(5)5167(5)58.0(13)c81001(5)6986(5)6771(5)57.1(12)c388481(5)6386(5)4415(5)51.8(12)c234057(6)2625(6)4312(5)70.8(15)c407634(6)7882(6)887(5)60.1(13)c174243(6)3641(6)8995(6)67.1(16)c2-706(6)10229(6)8011(5)61.3(14)c485436(6)7790(5)5213(6)75.3(15)c477008(5)8083(5)4823(5)57.3(13)c7357(6)8178(5)6432(6)62.8(14)c2911615(9)-2201(9)1989(10)102(3)c5-2364(7)12341(6)7181(7)75.4(17)n612158(7)-1341(7)3399(7)101(2)c4-1580(7)12266(7)8041(6)76.8(17)c443942(6)7268(6)-236(6)67.4(16)c349370(5)4183(5)2302(4)50.9(12)c2810776(8)-1146(8)1577(7)87(2)c465696(6)8637(5)-274(5)55.9(13)c185428(6)3801(6)9379(6)72.0(17)c1-1548(6)10380(5)7151(5)55.8(12)c3211169(7)2719(7)2022(6)72.8(17)c3110575(7)1732(6)3028(5)64.7(15)c416282(5)7645(5)579(4)50.7(12)c244458(8)1956(11)3447(7)116(3)c3012262(10)-2294(9)2895(10)108(3)o104974(11)6061(7)4658(8)178(4)c505087(8)6417(7)5478(7)95(2)
[0114]
[表7]原子xyzu(eq)o95280(20)5578(9)6379(9)328(10)
c494896(8)8186(9)6272(10)121(3)
[0115]
继而,将氢原子的原子座标示于表8~表9。此处,u(iso)意指各向同性温度因子。另外,表8~9的氢原子的编号是与键合的非氢原子的编号相关联而添加的。[表8]原子xyzu(eq)h89346.775068.73913.7861h213688.72574.0510586.5558h3605.175124.266028.8258h11-2369.747373.128382.175h2710069.67531.081757.5575h15a2067.681310.629351.8161h15b1497.972573.949556.7361h357956.483311.883730.7672h205667.21833.8211271.2870h434035.485405.74950.9593h10a-1260.545381.39009.9773h3a-297.711150.919057.2586h426008.865728.151629.3982h3310953.54619.29949.3376h454185.239145.91-1286.0674h368922.521244.534318.2980h22a2066.941977.024712.7870h22b2984.221087.665637.7470h8a1852.436897.226360.7269h233869.033552.193869.5185h40a8359.197555.79439.3372h40b7769.658823.57624.3672h173883.024354.248424.6481h2-128.049513.178265.2374h484971.948339.684545.790h47a7150.329026.024438.2569h47b7459.017775.335525.2469h7769.298894.745794.6275h2911730.8-2832.71649.99122h5a-2923.1513066.866898.9890h4-1599.1712938.918326.2392h2810297.82-1073.81960.63105h466096.119469.21-582.5767h185876.374610.889061.1186
h3211978.742566.141591.7287
[0116]
[表9]原子xyzu(eq)h24a5221.962413.892873.1175h24b3696.381963.343045.27175h24c4710.681064.73868.09175h3012805.06-3025.753202.32130h104972.255256.084903.89267h49a5188.957556.536980.6182h49b3912.18211.166373.01182h49c5249.849039.796125.23182h56580(60)1560(80)5680(70)140(30)
[0117]
进而,将原子间键合距离(单位:埃)示于表10~表11。[表10]原子原子长度/a原子原子长度/acl2c441.744(6)c26c271.336(8)cl1c191.746(5)c26n61.337(8)n5c121.387(6)n10c371.403(6)n5c151.474(6)n10c401.473(7)n5c141.390(6)c27c281.368(9)n9c391.389(6)n7c371.271(7)n9c381.390(6)n7c341.423(6)n9c471.448(7)c19c201.381(8)n2c91.405(6)c19c181.370(8)n2c121.262(6)c35c361.381(8)c16c211.388(6)c35c341.380(8)c16c151.513(7)c43c421.396(9)c16c171.370(8)c43c441.383(8)o5c251.288(5)c3c21.384(8)n8c371.371(6)c3c41.329(9)n8c381.344(6)c42c411.387(8)c9c101.391(7)c25c231.527(7)c9c81.392(8)c33c341.388(7)o2c131.216(6)c33c321.392(8)o6c261.367(7)c45c441.339(9)o6c311.386(7)c45c461.375(8)c12n31.391(6)c36c311.385(9)c21c201.382(7)c22c231.514(8)o3c141.194(5)c8c71.369(7)
o7c381.210(6)c23c241.501(9)o8c391.222(6)c40c411.505(8)n3c131.364(6)c17c181.372(8)c6o11.412(6)c2c11.401(8)c6c111.368(9)c48c471.563(8)c6c71.365(8)c48c501.467(8)o4c251.191(6)c48c491.532(10)o1c11.364(6)c29c281.368(12)n4c131.375(5)c29c301.337(13)n4c141.388(6)c5c41.381(9)n4c221.481(6)n6c301.398(12)
[0118]
[表11]原子原子长度/a原子原子长度/an1c51.331(7)c46c411.371(7)n1c11.309(7)c32c311.366(10)c11c101.376(8)o10c501.231(10)c39n101.359(7)c50o91.178(13)
[0119]
关于式(i)所示的化合物的无水物i型晶体,于非对称单元中,式(i)所示的化合物存在2个分子。将式(i)所示的化合物的分子结构图分别示于图7及图8中。需要说明的是,表5~表7及表10~表11中的非氢原子的编号分别与图7及图8中所记载的编号对应。
[0120]
如表10~表11中所记载,c12-n2的键合距离显示约c37-n7的键合距离显示约由于c12-n2的键合距离及c37-n7的键合距离短于c12-n3的键合距离(约)及c37-n8的键合距离(约),因此无水物i型晶体中的式(i)所示的化合物均鉴定为亚氨基结构:[化学式49]实施例4
[0121]
(通过添加剂而得的氮杂-迈克尔加成反应的促进效果)[化学式50]
与上述流程图类似的反应于tetrahedron asymmetry,vol.7,no.3,pp.699-708,1996(非专利文献16)中有记载。该文献中,通过使用甲醇作为反应溶剂进行9天加热回流而获得氮杂-迈克尔加成反应的产物(产率74%,1∶1非镜像异构混合物)。由于作为原料的甲基丙烯酸甲酯(化合物2)为用作聚合物合成的原料的化合物,若长时间于高温下反应有会聚合之虞,因此本反应条件不适合于工业制造方法。对于此,判明:若于上述氮杂-迈克尔加成反应中使用氯化锂、氯化钙、氯化镁、溴化锂、对甲苯磺酸、甲磺酸或三氟甲磺酸作为添加剂,则如下述表12所示,可促进反应。[表12]以与实施例1相同的方式于上述表12的各条件下进行反应。一面搅拌各反应溶液一面取样,称取约100mg并加入液相色谱用甲醇稀释成100ml后,注入10μl,测定hplc(方法a)。*conv.(%)通过下式进行计算。[数学式1]此处,a1、a3'、a3”表示hplc测定中的各峰面积。
a1:化合物(1)a3':化合物(3')a3”:化合物(3”)需要说明的是,化合物(3”)为以下所示的化学结构式。[化学式51]迄今为止,只知晓如非专利文献16记载中所记载的进行9天加热回流的反应条件,通过在所述添加剂存在下进行反应可促进反应,反应会于1小时~7小时的短时间内完成,因此本制造方法可以说是工业上优异的制造方法。
[0122]
再者,非专利文献16中,是在(s)-1-苯基乙基胺((s)-α-甲基苄胺)与甲基丙烯酸甲酯的氮杂-迈克尔加成反应完成后,加入对甲苯磺酸而获得对甲苯磺酸盐。相对于此,本技术中,是在(r)-1-苯基乙基胺((r)-(+)-1-苯基乙基胺)与甲基丙烯酸甲酯的氮杂-迈克尔加成反应完成后,加入对甲苯磺酸而获得对甲苯磺酸。即,由于非专利文献16中获得的是本技术的式(iv-b):[化学式52]的镜像异构体的对甲苯磺酸盐,因此本技术发明未记载于非专利文献16中。实施例5
[0123]
(化合物(4')的硫酸盐与盐酸盐的分离产率比较)[化学式53]与上述实施例2中记载的制造方法相同,在去除钯碳后的滤液中加入甲醇及4mol/l盐酸-乙酸乙酯溶液。重复进行加入作为晶析溶剂的乙酸乙酯而进行减压蒸馏去除的操作,将溶剂置换为乙酸乙酯并冷却至0℃,滤取所析出的固体,由此获得化合物(4')的盐酸盐。化合物(4')的硫酸盐是依照上述实施例2而进行合成的。此处,对化合物(4')的盐酸盐或化合物(4')的硫酸盐的分离产率进行测定,及对滤取所析出的固体后的各滤液,称取约200mg并加入液相色谱用乙腈15ml及三乙胺45μl之后,加入苯甲酰氯15μl并加入液相色谱用乙腈稀释为20ml后,注入10μl而测定hplc(方法b)。另外称取作为浓度计算用基准溶液的化合物(4')的盐酸盐或化合物(4')的硫酸盐的分离固体约10mg并加入液相色谱用乙腈15ml及三乙胺45μl后,加入苯甲酰氯15μl并加入液相
色谱用乙腈稀释为20ml,其后注入10μl而测定hplc(方法b),通过下式计算出滤液中所含的化合物(4')的浓度。[数学式2]此处,ms、m
l
、as、a
l
表示hplc测定中的各峰面积。ms:化合物(4')的盐酸盐或化合物(4')的硫酸盐的分离固体的hplc测定中的称取量(mg)m
l
:滤液的hplc测定中的称取量(mg)as:化合物(4')的盐酸盐或化合物(4')的硫酸盐的分离固体的hplc测定中的峰面积a
l
:滤液的hplc测定中的峰面积[表13]条目盐分离产率(%)滤液损耗(%)晶析溶剂1盐酸盐88.0-acoet2盐酸盐83.07.1acoet3盐酸盐79.010.3acoet4盐酸盐82.96.2acoet5盐酸盐83.46.5acoet6盐酸盐77.03.0acoet7硫酸盐92.42.5mecn8硫酸盐94.03.0mecn9硫酸盐91.91.8mecn10硫酸盐93.43.0mecn如根据表13所明了,在以盐酸盐的形式对化合物(4')进行分离的情形(条目1~6)时,分离产率为约77%~约88%,存在变动。另外,确认到滤液中所溶出的游离体(化合物(4'))为约3%~约10%。相对于此,明确:在以硫酸盐的形式对化合物(4')进行分离的情形(条目7~10)时,以分离产率为约92%~约94%的高产率而获得。另外,滤液中所溶出的游离体的比率为约2~约3%。根据以上结果,以硫酸盐的形式制造化合物(4')的方法由于滤液中游离体溶出的量较少、制造时的损耗较小,因此可以说是工业上优异的制造方法。实施例6
[0124]
(外消旋化抑制效果)[化学式54]
专利文献6、7、8及9中公开了将酯进行水解而获得羧酸的制造方法,作为溶剂,使用的是二噁烷、thf、dmso、meoh-thf混合液、thf-etoh-水混合液、meoh-水混合液、meoh-thf-水混合液等。相对于此,判明:若于上述水解反应中使用异丙醇、thf或t-buoh作为反应溶剂,则如下述表14所示,可抑制产物之外消旋化。[表14]与实施例3相同地于上述表的条件下进行反应。对各反应液,称取约100mg并加入液相色谱用甲醇稀释为20ml后注入10μl,测定hplc(方法c)。*化合物(9)的r体量为通过下式进行计算。单位表示峰面积(%)。[数学式3]**式(i)所示的化合物的r体量为通过下式进行计算。单位表示峰面积(%)。[数学式4]
再者,化合物(9)的r体及式(i)所示的化合物的r体的结构式如以下所示。再者,化合物(9)的r体及式(i)所示的化合物的r体的分子结构(氨基体/亚氨基体)未确定。[化学式55]如上述表14所记载,可知:在使用甲醇作为反应溶剂的情形时,产生外消旋化,光学异构体的量自0.43%增加至0.93%。另外,在乙醇的情形时,亦相同,自0.43%增加至约0.7%。相对于此,于使用异丙醇、四氢呋喃及叔丁醇的情形时,增加至约0.5%。根据以上结果,明确:与使用甲醇及乙醇作为反应溶剂的情形相比,于使用异丙醇、四氢呋喃及叔丁醇的情形时,为自作为起始原料的化合物(9)中起初所含有的r体的比率稍有增加的程度,可抑制外消旋化。因此,本制造方法可以说是工业上优异的制造方法。实施例7
[0125]
[1]式(i)所示的化合物的二水合物晶体的合成在式(i)所示的化合物的无水物i型晶体(50.00g,98.43mmol)中加入2-丙醇(314.02g)、水(150.00g)、及48%氢氧化钠(20.51g,246.1mmol)进行溶解。于所获得的溶液中加入35%盐酸(25.64g,246.1mmol)及式(i)所示的化合物的二水合物的种晶(50.00mg)后,在室温下搅拌1小时,加入水(250.00g)搅拌2小时。将所获得的浆料冷却至5℃并进行过滤,由此获得式(i)所示的化合物的二水合物晶体(49.23g)。为二水合物晶体是利用示差热-热重量同步测定(tg/dta)、水分吸附脱附测定(dvs)及粉末x射线衍射测定确认到的。
[0126]
将式(i)所示的化合物的二水合物晶体的粉末x射线衍射图案示于图4(方法1)。衍射角度(2θ):5.7
°±
0.2
°
、7.7
°±
0.2
°
、11.8
°±
0.2
°
、15.2
°±
0.2
°
、17.7
°±
0.2
°
、20.6
°±
0.2
°
、20.8
°±
0.2
°
、26.5
°±
0.2
°
、27.1
°±
0.2
°
及29.1
°±
0.2
°
处的峰作为式(i)所示的化合物的二水合物晶体尤其具有特征性。
[0127]
将式(i)所示的化合物的二水合物晶体的拉曼光谱的结果示于图6(方法2)。于871cm-1
±
2cm-1
、996cm-1
±
2cm-1
、1093cm-1
±
2cm-1
、1114cm-1
±
2cm-1
、1234cm-1
±
2cm-1
、1248cm-1
±
2cm-1
、1340cm-1
±
2cm-1
、1577cm-1
±
2cm-1
、1603cm-1
±
2cm-1
、1662cm-1
±
2cm-1
、1738cm-1
±
2cm-1
、2971cm-1
±
2cm-1
、3073cm-1
±
2cm-1
处确认到主要吸收峰。
[0128]
在一个实施方案中,式(i)所示的化合物的二水合物晶体于871cm-1
±
2cm-1
、996cm-1
±
2cm-1
、1114cm-1
±
2cm-1
、1234cm-1
±
2cm-1
、1340cm-1
±
2cm-1
及1577cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的二水合物晶体于871cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的二水合物晶体于996cm-1
±
2cm-1
处具有
吸收峰。在一个实施方案中,式(i)所示的化合物的二水合物晶体于1114cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的二水合物晶体于1234cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的二水合物晶体于1340cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的二水合物晶体于1577cm-1
±
2cm-1
处具有吸收峰。在一个实施方案中,式(i)所示的化合物的二水合物晶体具有选自由871cm-1
±
2cm-1
的吸收峰、996cm-1
±
2cm-1
的吸收峰、1114cm-1
±
2cm-1
的吸收峰、1234cm-1
±
2cm-1
的吸收峰、1340cm-1
±
2cm-1
的吸收峰及1577cm-1
±
2cm-1
的吸收峰所组成的组的1个以上吸收峰。
[0129]
将式(i)所示的化合物的二水合物晶体的示差热热重量同步测定(tg/dta)的结果示于图5。其结果确认到:自约55℃至约85℃,伴随吸热峰,重量减少了6.4%。由在式(i)所示的化合物的二水合物晶体的水分含量的理论值为6.6%,因此确认为式(i)所示的化合物的二水合物晶体。
[0130]
[2]式(i)所示的化合物的二水合物的种晶的合成于化合物(9)(70.00g,134.1mmol)中加入2-丙醇(109.91g)、水(63.02g)、及48%氢氧化钠(27.95g,335.4mmol),于25℃下搅拌4小时。于所获得的反应溶液中加入2-丙醇(16.49g)、甲醇(127.43g)、及水(217.00g)后,以25℃加入甲酸(9.88g,215mmol),于25℃下搅拌35分钟。于所获得的浆料中以25℃滴加将甲酸(8.64g,188mmol)与水(70.00g)混合所制备的甲酸水溶液后,加入水(7.00g)及甲醇(27.70g)。将浆料进行过滤,由此获得式(i)所示的化合物的二水合物的种晶(64.04g)。
[0131]
(参考例4)(专利文献9记载的化合物i-127的合成)如上所述,专利文献9中,并未具体地记载化合物i-127的各制造步骤。与化合物i-127的类似化合物(专利文献9的参考例3)同样地合成化合物i-127。以下仅公开最终步骤。[化学式56]将(s,e)-3-(3-(4-氯苄基)-2,6-二氧代-4-((4-(吡啶-2-基氧基)苯基)亚氨基)-1,3,5-三嗪-1-基)-2-甲基丙酸甲酯(0.365g,0.7mmol)、meoh(1ml)、thf(1ml)、h2o(1ml)、及4mol/l-lioh水溶液(0.7ml,2.80mmol)加以混合,在室温下搅拌2小时。将反应液与半饱和盐水(100ml)加入至5%柠檬酸溶液中,利用乙酸乙酯(100ml)进行萃取,并利用半饱和盐水(100ml)对有机层进行洗净。利用硫酸镁使有机层干燥,其后进行减压蒸馏去除。利用柱
色谱法(乙酸乙酯/己烷=50:50~乙酸乙酯/己烷=80:20)对所获得的残渣进行纯化,其后利用乙酸乙酯/己烷混合溶剂进行再晶体,滤取后于80℃下减压干燥3小时,获得白色粉末。1h nmr(dmso-d6)δ:0.86(1.5h,t,j=6.8hz,己烷),1.03(3h,d,j=6.8hz),1.18(0.75h,t,j=7.4hz,acoet),1.25(2h,brs,己烷),1.99(0.75h,s,acoet),2.76(1h,t,j=7.2hz),3.81(1h,t,j=10.5hz),3.95(1h,t,j=10.2hz),4.03(0.5h,q,j=7.4hz,acoet),5.29(2h,s),7.02-7.12(4h,m),7.36-7.45(6h,m),7.85(1h,t,j=7.5hz),8.16(1h,s),9.34(1h,brs).lc/ms m/z 508[m+]保留时间2.02min此处,lc/ms为专利文献9的方法1的条件下进行测定。
[0132]
将1h-nmr的结果示于图9。于nmr的光谱图中,确认到乙酸乙酯及己烷的峰。由于所获得的晶体于80℃下减压干燥过3小时,因此推测并非晶体表面的附着溶剂,而是晶格所内包的乙酸乙酯及己烷。另外,由于在tg/dta(图13)中确认到伴随吸热的重量减少,因此提示为乙酸乙酯及己烷的溶剂合物晶体。进而,根据1h-nmr的积分比,式:[化学式57]所表示的化合物、乙酸乙酯、己烷的存在摩尔比为约4:1:1。因此,推定:专利文献9中记载的化合物i-127获得了式:[化学式58]所表示的化合物的乙酸乙酯/己烷溶剂合物晶体(但是,关于分子结构(氨基体/亚氨基体),不详)。
[0133]
继而,将所获得的白色粉末的粉末x射线衍射的图案示于图10(方法2)。于粉末x射线衍射光谱中,于衍射角度(2θ):8.3
°±
0.2
°
、10.8
°±
0.2
°
、13.4
°±
0.2
°
、17.9
°±
0.2
°
、19.4
°±
0.2
°
、21.7
°±
0.2
°
、25.6
°±
0.2
°
处确认到峰。
[0134]
另外,将所获得的晶体的拉曼光谱的结果示于图11(方法2)。于821cm-1
±
2cm-1
、856cm-1
±
2cm-1
、893cm-1
±
2cm-1
、1002cm-1
±
2cm-1
、1094cm-1
±
2cm-1
、1221cm-1
±
2cm-1
、1269cm-1
±
2cm-1
、1575cm-1
±
2cm-1
、1604cm-1
±
2cm-1
、1614cm-1
±
2cm-1
、1649cm-1
±
2cm-1
、1725cm-1
±
2cm-1
及3073cm-1
±
2cm-1
处确认到主要吸收峰。于821cm-1
±
2cm-1
、1002cm-1
±
2cm-1
、1269cm-1
±
2cm-1
、1575cm-1
±
2cm-1
、1614cm-1
±
2cm-1
、1649cm-1
±
2cm-1
及1725cm-1
±
2cm-1
处确认到主要吸收峰。于1575cm-1
±
2cm-1
、1614cm-1
±
2cm-1
及1649cm-1
±
2cm-1
处确认到主要吸收峰。于1614cm-1
±
2cm-1
及1649cm-1
±
2cm-1
处确认到主要吸收峰。实施例8
[0135]
(式(i)所示的化合物的无水物ii型晶体的合成)称取式(i)所示的化合物的无水物i型晶体(约10mg)至小瓶(vial)中,添加200μl的chcl3。于25℃下以400rpm利用磁搅拌器进行搅拌(整夜运转)。7天后进行过滤,并利用粉末x射线衍射测定对所获得的粉末进行确认。将式(i)所示的化合物的无水物ii型晶体的粉末x射线衍射图案示于图12(方法3)。于粉末x射线衍射光谱中,于衍射角度(2θ):8.5
°±
0.2
°
、12.1
°±
0.2
°
、16.4
°±
0.2
°
、17.1
°±
0.2
°
、18.1
°±
0.2
°
、18.5
°±
0.2
°
、20.3
°±
0.2
°
、23.0
°±
0.2
°
及24.7
°±
0.2
°
处确认到峰。于上述粉末x射线衍射光谱中,于衍射角度(2θ):8.5
°±
0.2
°
、12.1
°±
0.2
°
、16.4
°±
0.2
°
、17.1
°±
0.2
°
及18.1
°±
0.2
°
处确认到峰。实施例9
[0136]
(流动性试验(压缩度及hausner比))(测定方法)1)将约10g的试样平静地放入50ml量筒中,测定试样量。2)小心地刮平而不压密粉体层的上表面,缓慢地读取松体积(v0)。3)将量筒安装于粉末测试机(pt-x型,hosokawa micron)的支持台。4)将粉体试样振实10次、500次、1250次、2500次,读取对应的松体积(v
10
、v
500
、v
1250
、v
2500
)。5)于体积的差成为最小刻度(0.5ml)以下的时点结束,将最终的松体积作为最终振实体积(vf)。6)重复3次1)~5)进行测定,于压缩度及hausner比的算出中采用平均值。
[0137]
(压缩度及hausner比算出方法)压缩度=(v
0-vf)/v0×
100hausner比=v0/vf[0138]
(结果)将结果示于表15。[表15]
如根据表15所明了,判明:式(i)所示的化合物的无水物i型晶体其压缩度(%)最低,与其他2个晶体形式相比,流动性良好(参考:pmda资料;粉体的流动性;压缩度及hausner比测定法,表16中显示摘选)。[表16]已知:一般而言,于药品的固体制剂化过程中,药物用粉体的流动性较高能够稳定地进行制造。药品粉体的低流动性可能成为粉体加工装置例如打片机的漏斗中的桥接现象、片剂重量偏差增大的原因。生产稳定的药品粉体时,要求流动性较高的晶体形式,且判明,式(i)所示的化合物的无水物i型晶体于药品的制剂化过程中为特别优选的晶体形式。实施例10
[0139]
(基于ich编码q3c的残留溶剂量)己烷为应限制药品中的残留量的溶剂(2类),乙酸乙酯为低毒性的溶剂(3类)。因此,药品原体中的己烷的残留量需要调整至规定值以下。以下的表17中表示基于ich编码q3c的2类的溶剂。[表17]
己烷的pde(permitted daily exposure,每日允许暴露量)为2.9mg/天。于使用式(i)所示的化合物的乙酸乙酯/己烷溶剂合物晶体作为药品原体的情形时,根据投予量,存在己烷的pde成为限制值以上的情况。相对于此,判明:式(i)所示的化合物的无水物i型晶
体及二水合物晶体不含作为残留溶剂的己烷,作为用于药品原体的晶体形式优异。实施例11
[0140]
(固体稳定性试验)(测定方法)以2ml小瓶精密地称取试样5mg,分别各称取4份,盖上瓶盖,于特定温度下保存(密闭保存)。继而,于特定期间保存后取出各者,并评价稳定性(含量)。(保存条件)80℃栓紧(保存期间)1周(试液制备条件)于25ml量瓶中将各小瓶的试样洗出。溶解时的介质:乙腈/水=1/1(hplc测定条件)方法d(结果)将结果示于表18中。[表18]在任一晶体形式中,残存率均为99%以上,判明为稳定的晶体形式。式(i)所示的化合物的乙酸乙酯/己烷溶剂合物晶体生成了5种类似物。式(i)所示的化合物的无水物i型晶体生成了3种类似物,式(i)所示的化合物的二水合物晶体生成了2种类似物。根据以上,判明:式(i)所示的化合物的无水物i型晶体及二水合物晶体生成的类似物的种类较少。
[0141]
以下所示的制剂例仅为例示,并非意在对发明的范围进行任何限定。本发明的化合物可通过任意先前的路径、特别是经肠例如以经口且例如片剂或胶
囊剂的形态、或以非经口且例如注射液剂或悬浮剂的形态、于局部且例如以洗液剂、凝胶剂、软膏剂或乳霜剂的形态、或经鼻形态或栓剂形态以药物组合物的形式进行投予。一并包含至少1种药学上可接受的载体或稀释剂及游离形态或药学上可接受的盐的形态的本发明的化合物的药物组合物可利用先前的方法通过混合、造粒或涂布法来制造。例如,作为经口用组合物,可制成含有赋形剂、崩解剂、结合剂、润滑剂等及有效成分等的片剂、颗粒剂、胶囊剂。另外,作为注射用组合物,可制成溶液剂或悬浮剂,可进行杀菌,另外,亦可含有保存剂、稳定剂、缓冲剂等。工业实用性
[0142]
作为本发明的式(i)所示的化合物的晶体作为原料药有用。另外,包含式(i)所示的化合物的晶体的药物组合物作为慢性咳嗽的治疗剂或预防剂非常有用。进而,本发明作为制造式(i)所示的化合物的方法有用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1