制备苯并二氢吡喃化合物的方法与流程
制备苯并二氢吡喃化合物的方法
1.相关申请的交叉引用
2.该pct申请要求于2019.11.19提交的印度临时专利申请号 201921047127的优先权,其内容通过引用整体并入本文。
3.所有参考文献,包括但不限于专利、专利申请、非专利参考文献和本发明中引用的产品,均通过引用整体并入本文。
技术领域
4.本发明涉及一种用于制造取代的苯并二氢吡喃化合物(chromancompounds)的方法。更具体地,描述了钙敏感受体(casr)调节剂2-甲基-5
‑ꢀ
((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸(2
‑ꢀ
methyl-5-((2r,4s)-2-((((r)-1-(naphthalen-1-yl)ethyl)amino)methyl)chroman-4
‑ꢀ
yl)benzoic acid)、其中间体及其药学上可接受的盐的有效和安全合成。还描述了以经济上可规模化的方式纯化2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸、其中间体及其药学上可接受的盐的有效方法。本发明所述的步骤在不使用自燃试剂和不使用柱色谱纯化步骤的情况下进行。本发明还涉及用于合成的中间体。特别地,本发明涉及钙敏感受体(casr)调节剂2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基) 甲基)苯并二氢吡喃-4-基)苯甲酸、其中间体及其药学上可接受的盐的合成。
背景技术:
5.以下包括对理解本发明有用的信息。不是承认本文具体地或隐含地引用的任何信息、出版物或文献对于所描述或要求保护的发明是现有技术或必不可少的。本文提及的所有出版物和专利都通过引用整体并入本文。
6.钙敏感受体是c类g蛋白偶联受体(gpcr)。它通过调节甲状旁腺激素的循环水平在维持生理血清离子钙(ca
2+
)浓度中起主要作用。细胞胞外ca
2+ ([ca
2+
]o)是casr的主要生理配体。
[0007]
称为拟钙剂的gpcr的正向变构调节剂的小分子,可调节并改善受体对已经存在的细胞外离子钙环境的敏感性并减少pth分泌。gpcr的调节已被探索作为甲状旁腺功能亢进和与casr信号传导降低相关疾病的潜在疗法。西那卡塞(cinacalcet)是第一个被美国食品和药品管理局(fda)批准的 casr调节剂。可调节casr的其它分子也是已知的,如wo2013/124828中所述。
[0008]
wo2013/124828公开了用于casr调节的一系列取代的苯并二氢吡喃化合物。其中公开的一种具体化合物是2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基) 乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐(2-methyl-5-((2r,4s)-2
‑ꢀ
((((r)-1-(naphthalen-1-yl)ethyl)amino)methyl)chroman-4-yl)benzoic acidhydrochloride)。该申请还描述了合成这些取代的苯并二氢吡喃化合物的一般方法。该申请中描述的方法的缺点包括使用自燃试剂,这是危险的,因此对于大规模使用是不可行的,以及纯化技术例如快速色谱和用于
分离化合物的昂贵的手性色谱技术。该合成还包括苛刻的氢化反应条件,其是形成不需要的杂质的原因。
[0009]
鉴于上述情况,需要一种更有效的方法,该方法不仅更经济,而且同时使用危险性更小的试剂来制备2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基) 氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐。
[0010]
本发明克服了这些限制,由此2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸及其药学上可接受的盐以及合成中的中间体可以在工业规模上制备,无需使用复杂或冗长的纯化步骤,并且也具有高纯度。
技术实现要素:
[0011]
本文描述和要求保护的发明具有许多属性和方面,包括但不限于在本发明内容中阐述或描述或引用的那些。但这并不意味着是包括一切的,并且在此描述和要求保护的发明并不限于在本发明内容中确定的特征或实施例,或者不受在本发明内容中确定的特征或实施例的限制,本发明内容仅为了说明而不是限制的目的被包括在内。
[0012]
在一些方面,本发明提供了2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基) 氨基)甲基)苯并二氢吡喃-4-基)苯甲酸及其药学上可接受的盐以及合成中的中间体的合成路线,其可以在工业规模上制备,无需使用复杂或冗长的纯化步骤,并且纯度高,2-甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基) 苯并二氢吡喃-4-基)苯甲酸盐酸盐(2-methyl-5-((2r,4r)-2-((((r)-1
‑ꢀ
(naphthalen-1-yl)ethyl)amino)methyl)chroman-4-yl)benzoic acid hydrochloride) (化合物-b)杂质低于约1.0%,具体地2-甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基) 乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐杂质小于约0.5%,更特别地2-甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4
‑ꢀ
基)苯甲酸盐酸盐杂质小于约0.2%。
[0013]
本发明公开的工艺和方法是高性价比的,涉及使用温和且易于操作的试剂,因此即使当以大的工业规模制备化合物时也是有利的。而且,这些工艺和方法不使用复杂或冗长的纯化程序,但是能够合成高质量和高纯度的化合物,具有小于约1.0%的2-甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基)乙基) 氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐(化合物-b)杂质,具体地小于约0.5%的2-甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐杂质,更具体地小于约0.2%的2-甲基-5-((2r,4r)-2
‑ꢀ
((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐杂质。
[0014]
在一些方面,本发明有利地提供了制备2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸及其药学上可接受的盐的工艺。
[0015]
在一些方面,本发明提供了制备2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基) 乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸(化合物-4)的工艺。
[0016]
在一些方面,本发明提供了制备2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基) 乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐(化合物-a)的工艺。
[0017]
在一些方面,本发明提供了制备2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基) 乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐的工艺,该工艺不使用任何色谱步骤来
分离/纯化。
[0018]
在一些方面,本发明提供了制备2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基) 乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐(化合物-a)的工艺,该化合物含有低于约1.0%的2-甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐(化合物-b)杂质,特别是低于约0.5%的2-甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基) 苯甲酸(2-methyl-5-((2r,4r)-2-((((r)-1-(naphthalen-1-yl)ethyl)amino)methyl) chroman-4-yl)benzoic acid)杂质,更特别是低于约0.2%的2-甲基-5-((2r, 4r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸杂质。
[0019][0020]
在一些方面,本发明提供制备2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐(化合物-a)的工艺,该化合物含有低于约1.0%的2-甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基) 苯并二氢吡喃-4-基)苯甲酸盐酸盐(化合物-b)杂质,特别是低于约0.5%的2
‑ꢀ
甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸杂质,更特别是低于约0.2%的2-甲基-5-((2r,4r)-2-((((r)-1-(萘-1-基)乙基) 氨基)甲基)苯并二氢吡喃-4-基)苯甲酸杂质,其中使用了结晶过程。
[0021]
在一些方面,本发明提供了一种从5-((r)-2-(((叔丁氧基羰基)((r)-1-(萘
‑ꢀ
1-基)乙基)氨基)甲基)-2h-苯并吡喃-4-基)-2-甲基苯甲酸甲酯(methyl 5-((r)-2
‑ꢀ
(((tert-butoxycarbonyl)((r)-1-(naphthalen-1-yl)ethyl)amino)methyl)-2h
‑ꢀ
chromen-4-yl)-2-methylbenzoate)(化合物-1)开始,按照包括以下的步骤制备 2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐(化合物-a)的工艺:
[0022]
a)用pd/c和甲酸铵还原5-((r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基) 氨基)甲基)-2h-苯并吡喃-4-基)-2-甲基苯甲酸甲酯(化合物-1),得到5-((2r)
‑ꢀ
2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)-2-甲基苯甲酸甲酯(methyl 5-((2r)-2-(((tert-butoxycarbonyl)((r)-1-(naphthalen-1
‑ꢀ
yl)ethyl)amino)methyl)chroman-4-yl)-2-methylbenzoate)(化合物-2)
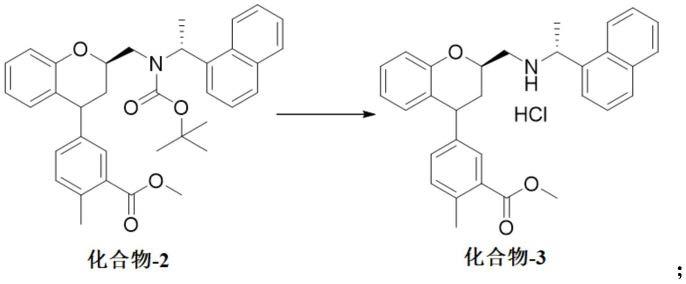
[0023][0024]
b)进行化合物-2的boc-脱保护反应,得到相应的氨基甲基2-甲基-5
‑ꢀ
((2r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸酯盐酸盐(amino methyl 2-methyl-5-((2r)-2-((((r)-1-(naphthalen-1-yl)ethyl)amino) methyl)chroman-4-yl)benzoate hydrochloride)(化合物-3)
[0025][0026]
c)水解化合物-3的酯基,并通过使用重结晶技术分离纯的非对映异构体,得到2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸(化合物-4)
[0027]
以及
[0028]
d)将化合物-4转化为其盐酸盐,2-甲基-5-((2r,4s)-2-(((((r)-1-(萘-1-基) 乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐
[0029][0030]
在一些方面,化合物-4可以转化为氢溴酸盐、氢碘酸盐、硫酸盐、硝酸盐、甲苯磺酸盐或碳酸盐。
[0031]
在一些方面,化合物-1中的双键被还原,得到化合物-2,其中所述反应通过使用甲酸铵(10eq)和5%pd/c(50%湿度和10%w/w载量)、10%pd/c 或2%pd/c在约33℃至约34℃温度下在乙酸乙酯-甲醇溶剂体系中进行。该转移氢化可以使用pd-c催化剂,使用甲酸盐作为氢源,例如甲酸铵或甲酸钠,在含水溶剂或有机溶剂中进行。本发明人设计了化合物-a的合成路线,包括甲酸盐作为氢源,使得合成路线能够安全地按比例放大,而不需要外源氢气作为还原剂。避免使用外源氢气还可以减少扩大规模所需的基础设施成本,否则就需要将与氢气爆炸相关的危险最小化。因此,本发明人已经认识到,本发明所述的不涉及使用外源氢气的合成路线具有显著的效益。
[0032]
在一些方面,化合物-1中双键还原得到化合物-2在约10℃至约50℃的温度下进行,更优选在约30℃至约33℃下进行。该反应可在任何合适的溶剂中进行,所述溶剂可包括或不包括:卤代烃、c
6-c
14
芳烃、c
1-c5醇、 c
2-c7酯、c
4-c7醚、c
1-c5羧酸、水或其合适的混合物。在一些方面,反应溶剂可以包括或不包括:水、甲醇、异丙醇、二氯甲烷、甲苯、乙酸乙酯、二乙醚及其组合。
[0033]
在一些方面,化合物-2的boc-去保护(boc-deprotection)反应通过在约 63℃回流下在甲醇中使用盐酸进行。在一些方面,盐酸的浓度是6n的 hcl水溶液。在一些方面,boc-去保护可以使用alcl3、三氟乙酸的二氯甲烷溶液或先三甲基碘化硅然后甲醇的顺序处理来进行。在一些方面,boc
‑ꢀ
去保护可以在阳离子清除剂存在下进行。阳离子清除剂可以包括或不包括苯甲醚或茴香硫醚。
[0034]
在一些方面,化合物-3的水解通过使用氢氧化钠在约55℃加热下在甲醇-四氢呋喃溶剂体系中进行。在一些方面,水解可以使用任何氢氧化物碱(例如氢氧化锂、氢氧化钾、氢氧化铯)或氯化锂进行,随后与所得的羧酸锂盐进行水反应生成羧酸。
[0035]
在一些方面,通过使用质子极性溶剂和非质子极性溶剂的溶剂混合物的重结晶技术从化合物-3的粗水解产物中分离非对映异构体的纯的化合物
‑ꢀ
4。在一些方面,质子极性溶剂可以包括或不包括:乙醇、甲醇、异丙醇或其组合。在一些方面,非质子极性溶剂可以包括或不包括二氯甲烷、二甲基甲酰胺、四氢呋喃或其组合。在一些方面,重结晶方法包括在溶剂中加热反应混合物,例如在溶剂-非溶剂的混合物中加热到55℃以上,并使溶液缓慢冷却到室温或更低,由此所需化合物(例如化合物4)的晶种优先结晶,而不需要的化合物(例如化合-3)基本上保留在溶液中。收集分离的基本上纯的产物(例如化合物-4),任选地随后用预冷却的溶剂-非溶剂溶液洗涤,得到基本上不含杂质的基本上纯化的化合物-4。
[0036]
在一些方面,从化合物-3的粗水解产物中分离非对映异构体的纯的化合物-4是通过使用乙醇:二氯甲烷溶剂混合物的重结晶技术进行的。在一些方面,乙醇与二氯甲烷的(v/v)比可以在1:5至5:1的范围内。
[0037]
在一些方面,化合物-4向化合物-a的转化通过使用盐酸进行酸中和来进行。在一些方面,盐酸是2n的hcl水溶液。
[0038]
在一些方面,本发明提供了一种制备5-((r)-2-(((叔丁氧基羰基)((r)-1
‑ꢀ
(萘-1-基)乙基)氨基)甲基)-2h-苯并吡喃-4-基)-2-甲基苯甲酸甲酯(化合物-1) 的工艺,该工艺包括以下步骤:
[0039]
a)在甲苯中用二(2-甲氧基乙氧基)氢化铝钠(sodium bis(2
‑ꢀ
methoxyethoxy)aluminium hydride)(例如以商品名vitridtm为人所知的二(2
‑ꢀ
甲氧基乙氧基)氢化铝钠)还原(r)-n-((r)-1-(萘-1-基)乙基)苯并二氢吡喃-2-甲酰胺((r)-n-((r)-1-(naphthalen-1-yl)ethyl)chroman-2-carboxamide)(化合物-5) 的酰胺基,然后用浓盐酸形成盐酸盐,得到(r)-n-((r)-苯并二氢吡喃-2-基甲基)-1-(萘-1-基)乙胺盐酸盐((r)-n-((r)-chroman-2-ylmethyl)-1-(naphthalen
‑ꢀ
1-yl)ethanamine hydrochloride)(化合物-6)
[0040][0041]
b)用boc酸酐(二碳酸二叔丁酯)和磷酸三钾保护化合物-6的游离氨基,得到((r)-苯并二氢吡喃-2-基甲基)((r)-1-(萘-1-基)乙基)氨基甲酸叔丁酯(tert
‑ꢀ
butyl((r)-chroman-2-ylmethyl)((r)-1-(naphthalen-1-yl)ethyl)carbamate)(化合物-7)
[0042][0043]
c)使用kmno4和mgso4氧化化合物-7,得到((r)-1-(萘-1-基)乙基)(((r)-4-氧代苯并二氢吡喃-2-基)甲基)氨基甲酸叔丁酯(tert-butyl((r)-1
‑ꢀ
(naphthalen-1-yl)ethyl)(((r)-4-oxochroman-2-yl)methyl)carbamate)化合物-8)
[0044][0045]
d)使化合物-8与三氟甲磺酸化剂(triflating agent)(其可包括或不包括n
‑ꢀ
苯
基-双(三氟甲磺酰胺)(n-phenyl-bis(trifluoromethanesulfonimide));三氟甲磺酸酐(trifluoromethanesulfonic anhydride);三氟甲磺酰氯(rifluoromethanesulfonyl chloride);三氟甲磺酸4-硝基苯酯(4-nitrophenyl trifluoromethanesulfonate)或1-(三氟甲磺酰基)咪唑)(1
‑ꢀ
(trifluoromethanesulfonyl)imidazole)))反应,得到(r)-2-(((叔丁氧基羰基)((r)
‑ꢀ
1-(萘-1-基)乙基)氨基)甲基)-2h-苯并吡喃-4-基三氟甲磺酸酯((r)-2-(((tert
‑ꢀ
butoxycarbonyl)((r)-1-(naphthalen-1-yl)ethyl)amino)methyl)-2h-chromen-4-yl trifluoromethanesulfonate)(化合物-9)
[0046]
以及
[0047]
e)在钯催化剂(其可包括或不包括:四(三苯基膦)合钯(palladium
‑ꢀ
tetrakis(triphenylphosphine));双(三苯基膦)二氯化钯 (ii)(palladium(ii)bis(triphenylphosphine)dichloride);钯(0)双(二亚苄基丙酮)(palladium(0)bis(dibenzylideneacetone));双(三苯基膦)二乙酸钯 (ii)(palladium(ii)bis(triphenylphosphine)diacetate);或[1,1'-双(二苯基膦基)二茂铁]二氯化钯(ii)([1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(ii))) 的存在下,将化合物-9与2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷
‑ꢀ
2-基)苯甲酸甲酯(methyl 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2
‑ꢀ
yl)benzoate)偶联,得到5-((r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基) 甲基)-2h-苯并吡喃-4-基)-2-甲基苯甲酸甲酯(化合物-1)
[0048][0049]
在一些方面,化合物-5的还原通过在甲苯(70%w/w)中使用二(2-甲氧基乙氧基)氢化铝钠)(例如以商品名vitrid
tm
为人所知的二(2-甲氧基乙氧基)氢化铝钠)进行。二(2-甲氧基乙氧基)氢化铝钠是非自燃还原剂。
附图说明
[0050]
附图形成本文说明书的一部分,并且被包括在内以进一步展示本文描述的实施例的某些方面。通过参考以下附图中的一个或多个并结合详细描述,可以更好地理解这些实施例。
[0051]
图1是通过本发明的代表性合成路线制备的化合物-a的粉末x-射线衍射图(x-轴
是2θ坐标)。
[0052]
图2是关于化合物-a和化合物-b的hplc色谱图实例,其通过本发明的代表性合成路线制备。相应的峰值表示于下表2中。
[0053]
图3是关于化合物-a和化合物-b的hplc色谱图实例,其通过本发明的代表性合成路线制备。相应的峰值表在下表3中给出。
具体实施方式
[0054]
应当理解,本文的过程、方法和/或系统不限于特定的合成过程、方法或系统、特定的组分或特定的组合物。还应理解,本文所用的术语仅是为了描述具体实施方案的目的,而不是旨在限制。
[0055]
定义
[0056]
式中使用的一般术语可以定义如下;然而,所陈述的含义不应被解释为限制术语本身的范围。
[0057]
如本文所用,变量的数值范围的描述旨在表达本发明可以用等于该范围内的任何值的变量来实施。因此,对于固有离散的变量,该变量可以等于数值范围的任何整数值,包括该范围的端点。类似地,对于固有地连续的变量,该变量可以等于数值范围的任何实数值,包括该范围的端点。作为示例,被描述为具有0和2之间的值的变量对于固有地离散的变量可以是0、1或2,并且对于固有地连续的变量可以是0.0、0.1、0.01、0.001或任何其他实数值。
[0058]
如本文所用,单数形式“一”、“一个”和“该”包括复数指代物,除非上下文另外明确指出。在本文中范围可以表示为从“约”一个特定值和/或到“约”另一个特定值。当表达这样的范围时,另一个实施方案包括从一个具体值和/或到另一个具体值。类似地,当通过使用先行词“约”将值表示为近似值时,应理解,特定值形成另一个实施方案。还应理解,每个范围的端点相对于另一个端点和独立于另一个端点都是有意义的。
[0059]
如本文所用,术语“约”旨在限定其修饰的数值,将这种值表示为误差范围内的变量。当没有叙述特定的误差范围,例如平均值的标准偏差时,术语“约”是指其所使用的数值的
±
10%。例如,“约50%”是指在45%至55%的范围内。
[0060]
本文所用的术语“烷基”是支链或非支链的烃基,例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、戊基、己基、庚基、辛基、壬基、癸基、十二烷基等。烷基也可以是取代或未取代的。除非另有说明,术语“烷基”包括取代的和未取代的烷基。烷基可以被一个或多个基团取代,所述基团包括但不限于烷氧基、烯基、炔基、环烷基、杂环烷基、芳基、杂芳基、醛、氨基、羧酸、酯、醚、卤化物、羟基、酮、硝基、甲硅烷基、硫代-氧代或硫醇。
[0061]
本文所用的术语“非对映异构体”是指彼此不是镜像的立体异构体。术语“异构体”是指分子式相同但原子的键合性质或顺序或原子在空间中的排列方式不同的化合物。在其原子空间排列方面不同的异构体称为“立体异构体”。非重叠镜像的非对映异构体称为“对映异构体”或有时称为“旋光异构体”。与四个不相同的取代基键合的碳原子被称为“手性中心”。具有一个手性中心且该手性中心具有相反手性的两种对映异构体形式的化合物被称为“外消旋混合物”。具有多于一个手性中心的化合物具有2n-1对映异构体对,其中n是手性中心的数目。具有一个以上手性中心的化合物可以作为单独的非对映异构体或作为非对映
异构体的混合物存在,称为“非对映异构体混合物”。当存在一个手性中心时,立体异构体可以通过该手性中心的绝对构型来表征。绝对构型是指连接到手性中心的取代基的空间排列。对映异构体可以通过于其手性中心的绝对构型来表征,并由cahn、ingold和prelog的r-和s-测序规则描述。立体化学命名法的惯例、立体化学的测定方法和立体异构体的分离方法是本领域公知的(例如,参见“advanced organic chemistry”,第4版,march,jerry,john wiley& sons,new york,1992)。
[0062]
本文所用的术语“药学上可接受的”是指其可用于制备药物组合物,该药物组合物通常是安全的、无毒的并且在生物学上或其它方面都不是有不良影响的,并且包括其对于兽医用途以及人类药物用途是可接受的。
[0063]
本文所用的术语“药学上可接受的盐”是指本发明的化合物的盐,其是药学上可接受的,如上所定义,并且其具有所需的药理学活性。这些盐包括与无机酸形成的酸加成盐,所述无机酸例如盐酸、氢溴酸、硫酸、硝酸、磷酸等;或与有机酸,例如乙酸、丙酸、己酸、庚酸、环戊烷丙酸、乙醇酸、丙酮酸、乳酸、丙二酸、琥珀酸、苹果酸、马来酸、富马酸、酒石酸、柠檬酸、苯甲酸、邻-(4-羟基苯甲酰基)苯甲酸(o-(4
‑ꢀ
hydroxybenzoyl)benzoic acid)、肉桂酸、扁桃酸、甲基磺酸、乙磺酸、1,2-乙二磺酸、2-羟基-乙磺酸、苯磺酸、对氯苯磺酸、2-萘磺酸、对甲苯磺酸、樟脑磺酸、4-甲基双环[2.2.2]辛-2-烯-1-甲酸(4-methylbicyclo[2.2.2]oct-2-ene
‑ꢀ
1-carboxylic acid)、葡庚糖酸、4,4'-亚甲基双(3-羟基-2-烯-1-甲酸)(4,4
′‑ꢀ
methylenebis(3-hydroxy-2-ene-1-carboxylic acid))、3-苯基丙酸、三甲基乙酸、叔丁基乙酸、月桂基硫酸、葡糖酸、谷氨酸、羟基萘甲酸、水杨酸、硬脂酸、粘康酸等反应。
[0064]
药学上可接受的盐还包括当存在的酸性质子能够与无机或有机碱反应时可以形成的碱加成盐。可接受的无机碱包括氢氧化钠、碳酸钠、氢氧化钾、氢氧化铝和氢氧化钙。可接受的有机碱包括乙醇胺、二乙醇胺、三乙醇胺、氨丁三醇、n-甲基葡糖胺等。
[0065]
如本文所用,术语“自燃”是指在空气存在下或在与空气接触后5分钟内自发点燃的物质。常用的还原剂通常是自燃的,包括lialh4、nah、氢化二异丁基铝、金属氢化物、三丁基锡、硼烷络合物(例如bh
3-thf)、氢气(与催化剂)等。如本文所用,“非自然”剂是指在空气存在下或在与空气接触后5分钟内不会自发点燃的物质。
[0066]
本文所用的术语“适当的结晶条件”是指选择的条件使得所需从化合物的混合物中结晶出来,优选化合物4从化合物3和化合物4的混合物中结晶。可用于进行该结晶的溶剂体系的实例包括但不限于醇和醇与一种或多种共溶剂的混合物,例如但不限于叔丁基甲基醚和二氯甲烷。选择乙醇与二氯甲烷的比例,以使所需化合物结晶。该比例可在约5:1至约1:5(v/v) 的范围内。合适的条件还可以包括加入酸。这些酸的实例包括盐酸。或者,也可以使用其它溶剂体系,例如质子溶剂和非质子溶剂的组合。在一些实施方案中,在溶液冷却时加入晶种以催化结晶过程。晶种可以包括或不包括硫酸钠、所需化合物的晶形或氯化钠。
[0067]
如本文所用,术语“质子溶剂”是指包含可离解的h
+
或能够形成氢键的基团(例如羟基或胺基)的溶剂。例如水、甲醇、乙醇、异丙醇、甲酸、氟化氢和氨。优选的质子溶剂包括醇,例如甲醇。
[0068]
如本文所用,术语“非质子溶剂”是指在所选反应的正常反应条件下不提供氢键的溶剂。例如hmpa、丙酮、thf(四氢呋喃)、乙醚、乙腈、 dmf(二甲基甲酰胺)、dmso(二甲基亚砜)、氯仿和dcm(二氯甲烷)。
[0069]
本文所述的化合物可通过合成有机化学工艺或方法制备。此外,在本文所述的方案中,在提及具体的碱、酸、试剂、溶剂、偶联剂等的情况下,应理解,也可以使用其它碱、酸、试剂、溶剂、偶联剂等,因此包括在本发明的范围内,除非另有说明。反应条件的变化,例如反应的温度和/或持续时间,如本领域已知的,也在本发明的范围内。除非另有说明,这些方案中描述的化合物的所有异构体也包括在本发明的范围内。本文提供的工艺如方案-1和方案-2中所述。除非另有说明,进行方案1或方案2的反应温度不是关键的。在某些实施方案中,当在反应中指示温度时,温度可以在约
±
0.1℃、约
±
0.5℃、约
±
1℃、约
±
5℃或约
±
10℃之间变化,这取决于在特定反应中使用哪种溶剂,最佳温度可以变化。在进行本文提供的反应时,反应物的加入速率和顺序都不是关键的,除非另有说明。除非另有说明,反应在环境大气压下进行。除非另有说明,反应物的精确量不是关键的。在一些实施方案中,反应物的量可以变化约10摩尔%或约10重量%。
[0070]
在一些实施方案中,本发明提供了化合物-a的合成路线,其是经济上可放大的,因此是商业上可制造的。本发明所述的化合物-a的合成路线不涉及柱层析纯化步骤,否则将导致实现商业量的化合物-a需要昂贵的制造成本。本发明人惊奇地开发了一种得到化合物-a的合成路线,该路线虽然包括了除wo2013/124828中所述步骤以外的其它步骤,但由于消除了损失产率的柱层析步骤,实际上得到了比参考文献的方法更高的总产率。本发明所述的合成路线也被设计为不使用自燃化合物和直接输入氢气,从而产生更安全的化合物-a的可制造方法。在一些实施方案中,使用二(2-甲氧基乙氧基)氢化铝钠(例如以商品名vitrid
tm
为人所知的二(2-甲氧基乙氧基)氢化铝钠)作为还原剂。二(2-甲氧基乙氧基)氢化铝钠是非自燃还原剂,其在功能上与氢化铝锂(氢化铝锂不用于本文所述的方法)相当。在一些实施方案中,使用含水/有机甲酸盐(例如甲酸铵)作为间接氢源代替氢气用于pd-c催化氢化。不受理论的约束,人们认为甲酸盐在溶液中离解产生氢,然后该氢暂时结合到pd催化剂表面上以充当氢化源。
[0071]
本发明提供了一种用于制备下式化合物-a的工艺(如方案-1所示):
[0072][0073]
其中所述工艺包括:
[0074]
1)在甲醇-乙酸乙酯溶剂体系中,在约33℃至约34℃加热条件下,用 5%pd/c和甲酸铵还原化合物-1,得到化合物-2;
[0075]
2)通过将化合物-2与6n的hcl的甲醇溶液回流,使化合物-2的boc
‑ꢀ
保护的胺基脱保护,得到化合物-3;
[0076]
3)通过在约55℃下用10n的naoh的甲醇-thf溶液加热化合物-3,水解化合物-3的酯基,得到粗化合物-4,进一步将得到的粗化合物用乙醇: dcm(5:1v/v)溶剂系统重结晶纯
化,然后用异丙醇重结晶,得到非对映异构体的纯的化合物-4;
[0077]
4)用2n的hcl水溶液将化合物-4转化为其盐酸盐(化合物-a)。
[0078]
方案1:
[0079][0080]
本发明提供了一种用于制备下式化合物-b的工艺(如方案-1a所示):
[0081][0082]
其中所述工艺包括以下步骤:
[0083]
1)通过在约55℃下用10n的naoh的甲醇-thf溶液加热化合物-3,水解化合物-3的酯基,得到粗化合物-4;
[0084]
2)用2n的hcl水溶液将化合物-4转化为其盐酸盐(化合物-b)。
[0085]
方案-1a:
[0086][0087]
本发明还提供了制备化合物-1的工艺(如方案-2所示),其包括:
[0088]
1)在甲醇-thf中在约85℃的加热条件下,通过在甲苯(70%w/w)中使用二(2-甲氧基乙氧基)氢化铝钠(例如以商品名vitrid
tm
为人所知的二(2-甲氧基乙氧基)氢化铝钠)还原化合物-5,得到化合物-5的还原产物,其在用浓盐酸处理后得到化合物-6;
[0089]
2)在磷酸三钾存在下,在dcm-水溶剂体系中,用boc酸酐保护化合物
ꢀ‑
6的游离氨基,得到化合物-7;
[0090]
3)在丙酮-水溶剂体系中,用高锰酸钾和硫酸镁氧化化合物-7,得到化合物-8;
[0091]
4)在双(三甲基甲硅烷基)氨基钾(khmds)和六甲基磷酰胺(hmpa)存在下,在-83℃下于thf中,使化合物-8与n-苯基-双(三氟甲磺酰亚胺) (phntf2)反应,得到化合物-9;
[0092]
5)在四(三苯基膦)钯(0)和磷酸三钾存在下,在thf中,在回流条件下,将化合物-9与2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯甲酸甲酯偶联,得到化合物-1。
[0093]
方案-2:
16小时。将反应物料冷却至15
±
5℃,在相同温度下加入饱和na2so3水溶液(0.84w/w)。将其搅拌30分钟。将硅藻土(1.0 w/w)加入上述反应物料中,在35
±
5℃下搅拌1小时,离心上述反应物,将滤饼加入含有丙酮(7.85w/w)的反应器中,回流3小时。过滤上述反应物料,浓缩至~6.0v。向其中加入饱和盐水溶液(1.20w/w)。用乙酸乙酯(10.0v
×
2) 萃取产物。分离有机层,用盐水洗涤(2.4w/w)。分离有机相,真空蒸发至~3.0v。将thf(3.0v)加入到上述溶液中并浓缩,得到((r)-1-(萘-1-基)乙基)(((r)-4-氧代苯并二氢吡喃-2-基)甲基)氨基甲酸叔丁酯。
[0124]
产率:99%
[0125]
质量:454.12[m+na]
[0126]1h nmr(400mhz,dmso-d6)δ:8.10
–
8.00(m,1h),7.97(dd,j=7.9, 1.6hz,1h),7.90(d,j=8.1hz,1h),7.69(d,j=7.2hz,1h),7.60
–
7.50(m, 4h),7.37(ddd,j=8.7,7.2,1.8hz,1h),6.92(td,j=7.6,1.0hz,1h),6.18(d,j =8.3hz,1h),3.66(tt,j=8.1,4.9hz,1h),3.40
–
3.21(m,3h),2.44(d,j= 14.4hz,1h),2.25(dd,j=17.0,3.2hz,1h),1.64(d,j=6.8hz,3h),1.48(d,j =12.2hz,9h).
[0127]
步骤-4:(r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)-2h-苯并吡喃-4-基三氟甲磺酸酯
[0128][0129]
在氮气下,向((r)-1-(萘-1-基)乙基)(((r)-4-氧代苯并二氢吡喃-2-基)甲基) 氨基甲酸叔丁酯(1.0eq)的thf(7.0v)溶液中加入hmpa(0.0015v)。在
‑ꢀ
83
±
5℃下,将双(三甲基甲硅烷基)氨基钾(potassium bis(trimethylsilyl)amide) (khmds)溶液(在thf中1m)(1.5eq)在1小时30分钟内滴加到上述溶液中。将上述反应物料在-83
±
5℃搅拌45分钟。在相同温度下,将n-苯基-双(三氟甲磺酰胺)(n-phenyl-bis(trifluoromethanesulfonimide))(phntf2)(1.5eq)的 thf(4.0v)溶液在3小时10分钟内滴加到上述溶液中,将其再搅拌30分钟。在-20
±
10℃下,用纯水(1.5v)淬灭反应,得到(r)-2-(((叔丁氧基羰基)((r)-1
‑ꢀ
(萘-1-基)乙基)氨基)甲基)-2h-苯并吡喃-4-基三氟甲磺酸酯,将其原样用于下一步骤。
[0130]
可选的分离步骤:反应完成后,用纯水(1.5v)在-20
±
10℃淬灭反应物。浓缩thf,然后用正己烷萃取产物(5v
×
3次),合并萃取液,用水(5v)洗涤,浓缩,得到(r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)-2h-苯并吡喃-4-基三氟甲烷磺酸酯。
[0131]
产率:84%
[0132]
质量:586.0[m+na]
[0133]1h nmr(400mhz,dmso-d6)δ:8.00(dt,j=6.9,3.5hz,2h),7.97
–
7.88 (m,1h),7.68(s,1h),7.57(ddd,j=8.7,6.9,3.0hz,3h),7.22(t,j=7.7hz, 1h),7.05(dd,j=7.7,1.6hz,1h),6.96(td,j=7.6,1.1hz,1h),6.45(s,1h), 6.09(s,1h),5.30(s,1h),3.95(s,
1h),1.63(d,j=6.9hz,3h),1.39(s,9h), 1.24(s,2h).
[0134]
步骤-5:5-((r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)-2h
‑ꢀ
苯并吡喃-4-基)-2-甲基苯甲酸甲酯
[0135][0136]
在氮气下,将(r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)
‑ꢀ
2h-苯并吡喃-4-基三氟甲磺酸酯的thf溶液(1.0eq)加入到反应器中。向上述溶液中加入2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯甲酸甲酯(0.95eq)和k3po4(1.5eq)。在氮气和环境温度下,将pd(pph3)4(1.2 mol%)加入到上述溶液中。将反应混合物加热至回流温度12-18小时。将反应物料冷却至环境温度,并向其中加入硅藻土(1w/w)、正庚烷(3.0v)和水 (1.0v)。过滤上述反应物料,分层,水相进一步用mtbe(2.0v)萃取。
[0137]
向合并的有机相中加入活性炭(0.2w/w)、硅胶(1.0w/w)和硅藻土(1.0 w/w)。将上述混合物在环境温度搅拌3小时。过滤上述混合物,真空蒸发反应物料至~2v。向上述反应物料中加入异丙醇(2.0v),蒸发至~2.0v,再次重复该共蒸馏过程。将上述物质冷却至5
±
5℃,在相同的温度下搅拌 4-8小时,过滤,得到5-((r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基) 甲基)-2h-苯并吡喃-4-基)-2-甲基苯甲酸甲酯,为湿滤饼。
[0138]
将上述滤饼在异丙醇(2.0v)中搅拌并加热至68
±
5℃,得到澄清溶液。然后将其冷却至15
±
5℃并在相同温度下搅拌16小时。过滤沉淀的固体,并用异丙醇(0.5v)洗涤固体。在40
±
5℃真空干燥上述固体,直到 lod≤0.5%。
[0139]
将上述固体加入到含有乙酸乙酯(3.15v)的反应器中。通过微孔过滤器过滤所得溶液。用纯水(1.5w/w)洗涤乙酸乙酯层10分钟。分离有机相,再次用纯水(1.5w/w)洗涤。分离有机层,在40
±
5℃真空蒸发至~1.5-2v。残余物与异丙醇(或乙醇)(1.57v)共蒸馏两次至~1.5-2v。向上述溶液中加入纯水(3.0w/w)。在40
±
5℃下,通过真空蒸发除去异丙醇至~3.5-4v。过滤沉淀的固体,用水(0.5v)洗涤。将这样得到的固体在真空烘箱中在45
±
5℃下干燥,直到lod≤0.5%,得到5-((r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)-2h-苯并吡喃-4-基)-2-甲基苯甲酸甲酯。
[0140]
产率:56.81%
[0141]
质量:586.44[m+na]
[0142]1h nmr(400mhz,dmso-d6)δ:8.08(d,j=8.0hz,1h),7.97(dd,j=8.1, 1.4hz,1h),7.91(d,j=8.2hz,1h),7.68(d,j=7.2hz,1h),7.65
–
7.49(m, 4h),7.35(d,j=7.9hz,1h),7.21(s,1h),7.10
–
7.02(m,1h),6.81
–
6.69(m, 2h),6.38(bs,1h),6.11(bs,1h),5.19(bs,1h),3.85(s,3h),3.75(bs,1h), 3.31(m,1h),2.53(s,3h),1.65(d,j=6.8hz,3h),1.50(bs,1h),1.32(bs,9h).
[0143]
可选步骤:在氮气下,向含(r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基) 氨
基)甲基)-2h-苯并吡喃-4-基三氟甲磺酸酯(110g,195mmol)的thf(体积: 500ml,比例:2.000)和水(体积:250ml,比例:1.000)的混合物的上述溶液中依次加入2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯甲酸甲酯(51.2g,185mmol)、磷酸三钾(91g,429mmol)和tetrakis(1.128g, 0.976mmol)。将混合物加热回流18小时。通过hplc监测反应进程。sm 完全消耗后,将该物质通过硅藻土垫过滤并减压浓缩。将残余物用水稀释,用正己烷萃取产物(5v
×
3次),合并的萃取液用水洗涤(5v),并减压浓缩。将上述残余物与乙醇(2v)共蒸馏,然后加入新鲜乙醇(4v),将得到的混合物温热,得到澄清溶液。冷却至室温并搅拌18小时,搅拌1小时,将物料冷却至0
±
5℃,过滤由此结晶的产物,并用冰冷的乙醇(1v)洗涤固体。将这样得到的固体在真空烘箱中于45
±
5℃下干燥,直到lod≤0.5%,得到5
‑ꢀ
((r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)-2h-苯并吡喃-4-基)
‑ꢀ
2-甲基苯甲酸甲酯。
[0144]
产率:82%
[0145]
质量:586.44[m+na]
[0146]1h nmr(400mhz,dmso-d6)δ:8.08(d,j=8.0hz,1h),7.97(dd,j=8.1, 1.4hz,1h),7.91(d,j=8.2hz,1h),7.68(d,j=7.2hz,1h),7.65
–
7.49(m, 4h),7.35(d,j=7.9hz,1h),7.21(s,1h),7.10
–
7.02(m,1h),6.81
–
6.69(m, 2h),6.38(bs,1h),6.11(bs,1h),5.19(bs,1h),3.85(s,3h),3.75(bs,1h), 3.31(m,1h),2.53(s,3h),1.65(d,j=6.8hz,3h),1.50(bs,1h),1.32(bs,9h).
[0147]
步骤-6:5-((2r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)-2-甲基苯甲酸甲酯
[0148][0149]
将甲酸铵(10.0eq)溶解在甲醇中,加热至33-34℃(6.0v),得到澄清溶液。将5-((r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)-2h-苯并吡喃-4-基)-2-甲基苯甲酸甲酯(1.0eq)溶解在乙酸乙酯(3.0v)中,加热至33
‑ꢀ
34℃,加入5%pd/c 50%湿产物(10%w/w g)。然后用加料漏斗将甲酸铵溶液在6小时内加入上述悬浮液中。将反应混合物在33-34℃加热2小时30 分钟。将反应混合物在4小时内冷却至20℃,并在20℃下搅拌9小时。通过gf/f玻璃微纤维过滤器滤出催化剂,用甲醇(1.0v)洗涤,然后用乙酸乙酯(2.0v)洗涤。将溶液在250mbar下连续地浓缩,并用乙酸乙酯稀释,以达到25/75的甲醇/乙酸乙酯摩尔比(nmr)。向由此获得的白色悬浮液中加入乙酸乙酯(4.0v),然后加入水(8.0v),使得两个均匀层易于分离。有机层用水(8.0v)洗涤,然后在250mbar下连续浓缩,用甲醇稀释以除去乙酸乙酯(nmr)。分离出中间体5-((2r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基) 氨基)甲基)苯并二氢吡喃-4-基)-2-甲基苯甲酸甲酯的甲醇溶液(~3.0v),其可用于下一步的合成。
[0150]
产率:100%
[0151]
质量:588.25[m+na]
[0152]1h nmr(400mhz,dmso-d6)δ:8.05(dd,j=8.1,1.5hz,2h),7.94(d,j =8.2hz,1h),7.72(d,j=7.1hz,1h),7.69
–
7.48(m,3h),7.34(d,j=2.0hz, 1h),7.24(d,j=7.9hz,1h),6.98(dd,j=7.8,2.0hz,1h),6.92(t,j=7.3hz, 1h),6.59(td,j=7.5,1.3hz,1h),6.33(s,1h),6.27(d,j=7.7hz,1h),6.14(s, 1h),3.83(s,3h),3.21(dd,j=14.4,5.9hz,2h),2.48(s,3h),1.66(s,3h),1.48 (bs,2h),1.37(bs,9h),1.24(s,2h).
[0153]
步骤-7:2-甲基-5-((2r)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸甲酯盐酸盐(methyl 2-methyl-5-((2r)-2-((((r)-1-(naphthalen-1
‑ꢀ
yl)ethyl)amino)methyl)chroman-4-yl)benzoate hydrochloride)
[0154][0155]
将5-((2r)-2-(((叔丁氧基羰基)((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)-2-甲基苯甲酸甲酯(300g,530mmol,1.0eq)的甲醇(1.2l,4.0v) 溶液加热至回流(63℃)。在63℃下,用滴液漏斗将6n的hcl水溶液(~ 352ml,2121mmol,4.0eq)在2小时内加入反应混合物中。将该溶液在 63℃下再搅拌一小时,接着以-10℃/h的速率冷却至20℃,然后在20℃下搅拌7小时。过滤白色悬浮液,首先用甲醇(225ml,0.75v)洗涤固体,然后用水[2
×
300ml(1v)]洗涤,得到2-甲基-5-((2r)-2-((((r)-1-(萘-1-基)乙基) 氨基)甲基)苯并二氢吡喃-4-基)苯甲酸甲酯盐酸盐,为白色湿盐酸盐。该产物准备用于下一步合成。
[0156]
产率:98%
[0157]
质量:466.12[mh+]
[0158]
1h nmr(400mhz,dmso-d6)δ:8.34
–
8.24(m,1h),7.96
–
7.88(m,1h), 7.78(d,j=8.1hz,1h),7.72s(dd,j=7.2,1.2hz,1h),7.63(d,j=1.3hz,1h), 7.56
–
7.44(m,3h),7.27(d,j=1.2hz,2h),7.06(tdd,j=7.1,2.0,1.0hz,1h), 6.79(dd,j=8.2,1.3hz,1h),6.71(td,j=7.5,1.3hz,1h),6.52(dt,j=7.7,1.4 hz,1h),4.67(q,j=6.5hz,1h),4.35
–
4.12(m,2h),3.78(s,3h),2.79(dd,j= 12.2,6.1hz,1h),2.64(dd,j=12.3,5.4hz,1h),2.49(s,3h),2.22(ddd,j= 13.5,5.8,1.7hz,1h),1.85
–
1.69(m,1h),1.41(d,j=6.5hz,3h).
[0159]
步骤-8:2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐
[0160][0161]
将2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃
‑ꢀ
4-基)苯甲酸甲酯盐酸盐(methyl-2-methyl-5-((2r,4s)-2-((((r)-1-(naphthalen-1
‑ꢀ
yl)ethyl)amino)methyl)chroman-4-yl)benzoate hydrochloride)(260g,518 mmol,1.0eq)溶解在甲醇(1.48l,5.7v)和四氢呋喃(1.48l,5.7v)的混合物中。将上述溶液加热至55℃,在20分钟内向其中加入10n的naoh(~ 260ml,2589mmol,5.0eq)。将澄清溶液在55℃搅拌2小时(ph 10)。将反应混合物冷却至30℃,并用水(1.82l,7.0v)稀释。缓慢加入2n的hcl 水溶液(1062ml,2124mmol,4.1eq)以调节ph至6-7。沉淀两性离子,将悬浮液冷却至20℃,并在该温度下搅拌30分钟。沙状固体易于过滤,首先用水[2
×
1300ml(5v)]洗涤,然后用etoh(520ml,2.0v)洗涤,接着用异丙醇(ipa)(260ml,1.0v)洗涤。将白色固体在40℃真空干燥20小时,得到2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基) 苯甲酸(粗产物,225g)。
[0162]
产率:96.15%
[0163]
纯度:83.79:15.57%
[0164]
纯化:将粗2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸(225g,498mmol,1.0eq)悬浮于5:1乙醇/二氯甲烷溶剂混合物(5.4l,24.0v)中。将悬浮液加热至剧烈回流(60℃)以使材料完全溶解。随后的重结晶在溶解结束之前开始。将悬浮液在60℃搅拌10分钟,然后以-20℃/h的速率冷却至20℃,过滤,首先用5:1乙醇/二氯甲烷溶剂混合物(2
×
675ml,3v)洗涤,然后用乙醇(225ml,1v)洗涤。将白色固体在40℃干燥过夜,得到2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸,为白色固体。
[0165]
产率:64.1%(150g)
[0166]
纯度:99.70:0.20%
[0167]
盐酸盐的制备:将由此得到的非对映异构体的纯的2-甲基-5-((2r,4s)
‑ꢀ
2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4-基)苯甲酸(150g,332 mmol,1.0eq)悬浮于水(2.55l,17v)中。在将反应物料加热至30℃后,快速加入2n的naoh水溶液(约300ml,598mmol,1.8eq)的溶液,使得化合物完全溶解。通过gf/a玻璃微纤维过滤器过滤溶液以除去任何固体杂质。然后,在相同温度下加入2n的hcl水溶液(665ml,1329mmol,4eq),诱导难以搅拌的大量白色固体沉淀。将反应物料在环境温度(22℃)下搅拌 20小时。过滤所得浆液,用水洗涤直至滤液的ph变为6[1500ml(10v),然后3
×
600ml(4v)]。在干燥箱中于40℃下干燥65小时后,以定量产率得到2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基)氨基)甲基)苯并二氢吡喃-4
‑ꢀ
基)苯甲酸盐酸盐白色固体。
[0168]
产率:89.53%
[0169]
纯度:99.63%
[0170]
质量:452.18[mh+]
[0171]
1h nmr(dmso-d6)δ:12.76(bs,1h),10.07(bs,1h),9.64(bs,1h),8.30 (d,j=8.4hz,1h),8.14
–
7.93(m,3h),7.73
–
7.56(m,4h),7.33
–
7.20(m, 2h),7.14(t,j=7.6hz,1h),6.87(dd,j=8.2,1.0hz,1h),6.79(td,j=7.6,1.1 hz,1h),6.57(d,j=7.7hz,1h),5.48(bs,1h),4.68(m,1h),4.29(dd,j=12.0, 5.7hz,1h),3.30(d,j=8.6hz,1h),3.20(d,j=12.8hz,1h),2.48(s,3h), 2.24(dd,j=12.7,5.3hz,1h),1.92(q,j=12.1hz,1h),1.77(d,j=6.6hz, 3h).
[0172]
ir(kbr,cm-1
):3057.55,2956.04,2876.08,2767.21,2681.29,2499.80, 2481.85,2298.48,2202.11,1711.42,1595.25,1579.33,1517.30,1497.94, 1483.60,1451.74,1400.13,1379.30,1362.67,1300.55,1279.31,1238.73, 1217.88,1187.99,1175.75,1118.41,1089.60,1072.72,1020.79,972.36,928.79, 913.23,892.94,860.86,797.19,780.99,745.77,704.12,667.76,611.33,571.04, 543.00,528.59,470.53,435.58,416.04,401.77.
[0173]
表1中的pxrd(x-射线粉末衍射图)峰由图1中所示的峰值得到。
[0174]
表1.非对映异构体的纯的2-甲基-5-((2r,4s)-2-((((r)-1-(萘-1-基)乙基) 氨基)甲基)苯并二氢吡喃-4-基)苯甲酸盐酸盐的pxrd峰(2-θ和相应的d间距)
[0175]
[0176][0177]
如前所述,杂质的量可以使用hplc(包括rp-hplc、hplc-ms/ms、 hplc-uv和iex)按照本领域已知的方法进行测量。例如,图2和3显示了实施例hplc色谱图,相应的峰值表分别列于表2和3中。每一峰值表揭示关于化合物a和化合物b的信息。
[0178]
表2-图2的峰值表
[0179][0180]
表3-图3的峰值表
[0181][0182]
下面是与图2和3相关的rp-hplc实验细节:
[0183]
rp-hplc:色谱柱:ymc triart(250
×
4.6)mm,5μm;
[0184]
运行时间(分钟):55.00;注射量:5.00μl;
[0185]
波长:wvl:220nm;流速:1.00ml/min;柱温40.0℃;
[0186]
流动相a:10mm nh4oac,ph-10.0:acn(90:10);以及
[0187]
流动相b:can:10mm nh4oac,ph-10.0(90:10)。
[0188]
在一些实施方案中,本发明提供:
[0189]
a1.一种化合物-a的合成方法,
[0190][0191]
其中所述合成不涉及自燃试剂。
[0192]
a2.如a1所述的方法,其中,所述合成包括以下步骤:
[0193]
1)在甲醇-乙酸乙酯溶剂体系中于33-34℃加热条件下,用5%pd/c和甲酸铵还原化合物-1,得到化合物-2
[0194][0195]
2)通过用6n的hcl的甲醇溶液回流化合物-2,使化合物-2的boc保护的胺基脱保护,得到化合物-3
[0196][0197]
3)通过在55℃下用naoh在甲醇-thf中加热化合物-3,水解化合物-3 的酯基,得到粗化合物-4,进一步使用乙醇:dcm(5:1)溶剂体系通过重结晶纯化得到的粗化合物,随后用异丙醇重结晶,得到非对映异构体的纯的化合物-4
[0198]
以及
[0199]
4)用2n的hcl水溶液将化合物-4转化为其盐酸盐(化合物-a)。
[0200][0201]
a3.一种合成化合物-1的方法,所述方法包括:
[0202]
a)在甲苯中使用二(2-甲氧基乙氧基)氢化铝钠(例如以商品名vitridtm为人所知的二(2-甲氧基乙氧基)氢化铝钠)还原化合物-5的酰胺基,随后使用浓盐酸形成盐酸盐,得到化合物-6
[0203][0204]
b)用boc酸酐(二碳酸二叔丁酯)和磷酸三钾保护化合物-6的游离氨基,得到化合物-7
[0205][0206]
c)用kmno4和mgso4氧化化合物-7得到化合物-8
[0207][0208]
d)使化合物-8与三氟甲磺酸化试剂反应,得到化合物-9
[0209]
以及
[0210]
e)在钯催化剂存在下,化合物-9与2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯甲酸甲酯偶联,得到化合物-1
[0211][0212]
a4.如a1所述的方法,其中,所述合成不包括柱层析纯化步骤来获得产物。
[0213]
a5.如a1所述的方法,其中,所述合成不涉及自燃试剂。
[0214]
a6.如a1所述的方法,其中,所述合成不涉及添加的氢气。
[0215]
a7.如a1所述的方法,其中,所述合成以1kg、10kg或100kg的规模进行。
[0216]
a8.如a4所述的方法,其中,所述合成不涉及手性柱色谱纯化方法得到产物。
[0217]
a9.如a4所述的方法,其中,所述合成不涉及自燃试剂。
[0218]
a10.一种获得化合物4的基本上非对映异构体的纯的组合物的方法,包括:在适当的结晶条件下从甲酯化合物3和化合物4的混合物中结晶所述化合物4。
[0219]
本文描述和要求保护的发明具有许多属性和实施例,包括但不限于本发明详细阐述或描述或引用的那些。但这并不意味着是包括一切的,并且在此描述和要求保护的发明并不限于本文详细公开的确定的特征或实施例,或者不受其限制,本文详细公开仅是为了说明而不是限制的目的而被包括。本领域普通技术人员将容易认识到,在不偏离本发明的范围的情况下,许多部件和参数可以在一定程度上改变或修改,或者可以用已知的等效物代替。应当理解,这些修改和等效物被包括在内,如同单独阐述一样。本发明还包括本说明书中单独或共同提及或指出的所有步骤、特征、组合物和化合物,以及任何两个或更多个所述步骤或特征的任何和所有组合。
[0220]
本文引用或提及的所有专利、出版物、科学文章、网站和其它文献和材料均表明本发明所属领域的技术人员的技术水平,且每个在此引用的文献和材料均以引用方式并入本文,其程度与通过单独引用的方式整体并入或在此整体阐述的程度相同。申请人保留将任何和所有来自任何此类专利、出版物、科学文章和其它参考材料或文献的材料和信息物理地并入本说明书的权利。本说明书中对任何申请、专利和出版物的引用不是也不应被视为承认或任何形式的暗示它们构成有效的现有技术或构成世界上任何国家的公知常识的一部分。
[0221]
本文所述的具体方法、工艺和化合物是优选实施方案的代表,并且是示例性的,而不是对本发明范围的限制。本领域技术人员在考虑本说明书时将想到的其它目的、实施方案和实施方案,并且这些目的、实施方案和实施方案被包括在如由权利要求的范围所限定的本发明的精神内。对于本领域技术人员来说,在不背离本发明的范围和精神的情况下,可以对本文公开的发明进行各种替换和修改是显而易见的。在此示例性描述的本发明可以在缺少任何一个或多个要素或一个或多个限制的情况下适当地实施,这些要素或限制在此没有作为必要的具体公开。因此,例如,在本文的每种情况下,在其实施方案或实施例中,任何术语“包括”、“基本上由
……
组成”和“由
……
组成”都可以替换为说明书中的其他两个术语中的任何一个。此外,术语“包括”、“包含”、“含有”等应被广义地理解而不是限制。本文示例性描述的方法和过程可以以不同的步骤顺序适当地实施,并且它们不必限于本文或权利要求中指出的步骤顺序。如本文和所附权利要求中所用的,单数形式“一”、“一个”和“该”也包括复数指代,除非上下文另外明确指出。在任何情况下,本发明都不能被解释为限于本文具体公开的具体实施例或实施方案或方法。在任何情况下,本发明均不得解释为受任何审查员或专利商标局的任何其他官员或雇员所作的任何声明的限制,除非该声明在申请人的书面回应文件中明确地且无条件或保留地明确采纳。此外,页面标题、标题等是为了增强读者对本文的理解,不应被视为限制本文的范围。本文所提及的本发明的实施例、实施例或组件的任何示例应被视为非限制性的。
[0222]
所采用的术语和表达方式被用作描述性的术语而非限制性的术语,并且使用这些术语和表达方式不旨在排除所示和所述特征或其部分的任何等同物,但是应当认识到,在所要求保护的本发明的范围内,各种修改是可能的。因此,应当理解,尽管已经通过优选实施方案和任选特征具体公开了本发明,但是本领域技术人员可以采用本文公开的概念的修改和变化,并且认为此类修改和变化在所附权利要求所限定的本发明的范围内。
[0223]
本发明已在本文中进行了广泛和一般性的描述。落入一般公开内容的每个较窄的种类和亚属分组也形成本发明的一部分。这包括本发明的一般性描述,附带条件或负面限制是从该属中除去任何有害物质,而不管所去除的物质是否在本文中具体叙述。
[0224]
其它实施例在以下权利要求范围内。另外,在本发明的特征或实施例以马库什的形式描述的情况下,本领域技术人员将认识到本发明也因此以马库什的任何单独成员或成员的分组的形式描述。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1