聚亚芳基硫醚的制造方法与流程
1.本发明涉及一种聚亚芳基硫醚的制造方法。
背景技术:
2.以聚亚苯基硫醚(以下,也称为“pps”)为代表的聚亚芳基硫醚(以下,也称为“pas”)是一种耐热性、耐化学品性、阻燃性、机械强度、电特性、尺寸稳定性等优异的工程塑料。pas能通过挤出成型、注塑成型、压缩成型等一般的熔融加工法成型为各种成型品、膜、片、纤维等,因此通用于电气设备、电子设备、汽车设备、包装材料等广泛的技术领域。
3.pas特别是在与汽车相关的领域中,多用作用于汽车的轻量化的金属替代物。在车载用途中,要求具有高韧性的高分子量pas。在专利文献1和专利文献2中,公开了制造粒状pas作为高分子量pas的方法。
4.现有技术文献
5.专利文献
6.专利文献1:日本特开2001-89569号公报
7.专利文献2:日本特开2004-51732号公报
技术实现要素:
8.发明要解决的技术问题
9.近年来,随着粒状pas的需求增加,要求粒状pas制造的进一步短时间化。但是,在现有制造方法中,以粒状pas的粒径控制、纯度提高为目的,在用于通过造粒形成粒状pas的冷却工序中,需要进行缓慢冷却,因此冷却工序需要长时间。
10.另一方面,已知当在聚合助剂的存在下进行pas的制造时,可在短时间内得到高分子量pas。但是,所得到的pas的平均粒径容易增大,其结果是,pas制造装置等的配管容易堵塞,此外,为了去除堵塞的pas,清洗配管需要大量时间。
11.本发明是鉴于上述问题而完成的,其目的在于,提供一种pas的制造方法,所述pas的制造方法能缩短用于形成粒状pas的冷却工序所花费的时间,并且,能抑制所得到的粒状pas的平均粒径增大。
12.技术方案
13.本发明人等发现通过如下方式能实现上述目的,从而完成了本发明,即,制造pas时,在选自羧酸盐等中的至少一种助剂的存在下,依次进行第一聚合工序、相分离剂添加工序、第二聚合工序、以及冷却工序,在冷却工序中,在规定的范围内的温度下向反应混合物添加冷却剂,并且将最大增粘温度下的冷却速度设为2.2℃/分钟以上且6.0℃/分钟以下。
14.本发明的pas的制造方法包括:第一聚合工序,在选自由羧酸盐、碱金属氯化物、有机磺酸盐、硫酸碱金属盐、碱土金属氧化物、碱金属磷酸盐、以及碱土金属磷酸盐构成的组中的至少一种助剂的存在下,对含有有机极性溶剂、硫源、水、多卤芳香族化合物、以及碱金属氢氧化物的混合物进行加热而引发聚合反应,生成含有多卤芳香族化合物的转化率为50
摩尔%以上的预聚物的反应混合物;相分离剂添加工序,在所述第一聚合工序后,向所述反应混合物添加相分离剂;第二聚合工序,在所述相分离剂添加工序后,继续聚合反应;以及冷却工序,在所述第二聚合工序后,对所述反应混合物进行冷却,在所述冷却工序中,在比所述最大增粘温度高5℃以上且小于250℃的温度下向所述反应混合物添加所述冷却剂,并且最大增粘温度下的冷却速度为2.2℃/分钟以上且6.0℃/分钟以下。
15.在本发明的pas的制造方法中,也可以是,在所述冷却工序中,向所述反应混合物添加冷却剂。
16.在本发明的pas的制造方法中,也可以是,所述冷却剂为水和/或冰。
17.在本发明的pas的制造方法中,也可以是,在所述最大增粘温度下,所述反应混合物中的冷却剂的含量相对于所述硫源1摩尔为2.7摩尔以上且6.0摩尔以下。
18.有益效果
19.根据本发明,能提供一种pas的制造方法,其能缩短用于形成粒状pas的冷却工序所花费的时间,并且,能抑制所得到的粒状pas的平均粒径增大。其结果是,pas制造装置等的配管变得不易堵塞,此外,去除堵塞的pas的必要性变低,因此能缩短清洗配管所花费的时间。根据以上,能整体上将粒状pas制造更短时间化。
具体实施方式
20.以下,对本发明的pas的制造方法的一个实施方式进行说明。就本实施方式中的pas的制造方法而言,作为必需的工序,包括:第一聚合工序、相分离剂添加工序、第二聚合工序、以及冷却工序。本实施方式中的pas的制造方法根据期望也可以包括:脱水工序、装料工序、后处理工序等。以下,对本发明所使用的各材料进行详细说明,并且对各工序进行详细说明。
21.(有机极性溶剂、硫源、多卤芳香族化合物、以及碱金属氢氧化物)
22.作为有机极性溶剂、硫源、多卤芳香族化合物、以及碱金属氢氧化物,可以使用在pas的制造中通常使用的物质。有机极性溶剂、硫源、多卤芳香族化合物、以及碱金属氢氧化物各自可以单独使用,若为能制造pas的组合,也可以混合两种以上使用。
23.作为有机极性溶剂,例如可列举出:有机酰胺溶剂;包含有机硫化合物的非质子性有机极性溶剂;包含环式有机磷化合物的非质子性有机极性溶剂。作为有机酰胺溶剂,可列举出:n,n-二甲基甲酰胺、n,n-二甲基乙酰胺等酰胺化合物;n-甲基-ε-己内酰胺等n-烷基己内酰胺化合物;n-甲基-2-吡咯烷酮(以下,也称为“nmp”)、n-环己基-2-吡咯烷酮等n-烷基吡咯烷酮化合物或n-环烷基吡咯烷酮化合物;1,3-二烷基-2-咪唑啉酮等n,n-二烷基咪唑啉酮化合物;四甲基脲等四烷基脲化合物;六甲基磷酸三酰胺等六烷基磷酸三酰胺化合物等。作为包含有机硫化合物的非质子性有机极性溶剂,可列举出二甲基亚砜、二苯砜等。作为包含环式有机磷化合物的非质子性有机极性溶剂,可列举出1-甲基-1-氧代正膦等。其中,从获取性、操作性等方面考虑,优选有机酰胺溶剂,更优选n-烷基吡咯烷酮化合物、n-环烷基吡咯烷酮化合物、n-烷基己内酰胺化合物以及n,n-二烷基咪唑啉酮化合物,更进一步优选nmp、n-甲基-ε-己内酰胺以及1,3-二烷基-2-咪唑啉酮,特别优选nmp。
24.从聚合反应的效率等观点考虑,有机极性溶剂的使用量相对于上述硫源1摩尔优选为1~30摩尔,更优选为3~15摩尔。
25.作为硫源,可列举出碱金属硫化物、碱金属氢硫化物、硫化氢,优选为碱金属硫化物和碱金属氢硫化物。就硫源而言,例如可以在水性浆料和水溶液中的任意种的状态下进行处理,从计量性、输送性等处理性的观点考虑,优选为水溶液的状态。作为碱金属硫化物,可列举出硫化锂、硫化钠、硫化钾、硫化铷、硫化铯。作为碱金属氢硫化物,可列举出氢硫化锂、氢硫化钠、氢硫化钾、氢硫化铷、氢硫化铯。
26.多卤芳香族化合物是指,与芳香环直接键合的两个以上氢原子被卤素原子取代而成的芳香族化合物,可以是与芳香环直接键合的两个氢原子被卤素原子取代而成的芳香族化合物(即,二卤芳香族化合物),也可以是与芳香环直接键合的三个以上氢原子被卤素原子取代而成的芳香族化合物(也称作“卤素取代数3以上的多卤芳香族化合物”)。
27.作为多卤芳香族化合物,例如可列举出:邻二卤苯、间二卤苯、对二卤苯、二卤甲苯、二卤萘、甲氧基-二卤苯、二卤联苯、二卤苯甲酸、二卤二苯基醚、二卤二苯砜、二卤二苯亚砜、二卤二苯酮等二卤芳香族化合物;1,2,3-三氯苯、1,2,4-三氯苯、1,3,5-三氯苯、六氯苯、1,2,3,4-四氯苯、1,2,4,5-四氯苯、1,3,5-三氯-2,4,6-三甲基苯、2,4,6-三氯甲苯、1,2,3-三氯萘、1,2,4-三氯萘、1,2,3,4-四氯萘、2,2’,4,4
’‑
四氯联苯、2,2’,4,4
’‑
四氯二苯甲酮、2,4,2
’‑
三氯二苯甲酮等卤素取代数3以上的多卤芳香族化合物。卤原子是指氟、氯、溴以及碘的各原子,多卤芳香族化合物中的两个以上的卤素原子可以相同或不同。其中,从获取性、反应性等方面考虑,优选对二卤苯、间二卤苯以及这两者的混合物,更优选对二卤苯,特别优选对二氯苯(以下,也称为“pdcb”)。
28.多卤芳香族化合物的使用量相对于硫源的装料量1摩尔优选为0.90~1.50摩尔,更优选为0.92~1.10摩尔,更进一步优选为0.95~1.05摩尔。若上述使用量在上述范围内,则不易产生分解反应,容易实施稳定的聚合反应,容易生成高分子量聚合物。
29.作为碱金属氢氧化物,例如可列举出氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铷、氢氧化铯。
30.(脱水工序)
31.脱水工序是在装料工序之前从包含含有有机极性溶剂、硫源以及碱金属氢氧化物的混合物的体系内将含水的馏出物的至少一部分排出至体系外的工序。硫源与多卤芳香族化合物的聚合反应根据存在于聚合反应体系的水分量而受到被促进或被阻碍等影响。因此,为了使上述水分量不阻碍聚合反应,优选的是,通过在聚合之前进行脱水处理来减少聚合反应体系内的水分量。
32.在脱水工序中,优选的是,通过在惰性气体气氛下的加热进行脱水。应该由脱水工序脱水的水分是指,在脱水工序中装入的各原料所含有的水、水性混合物的水介质、由各原料间的反应而副产的水等。
33.脱水工序中的加热温度只要为300℃以下就不特别限定,优选为100~250℃。加热时间优选为15分钟~24小时,更优选为30分钟~10小时。
34.在脱水工序中,脱水至水分量成为规定的范围内。即,在脱水工序中,理想的是,脱水至相对于装料混合物(后述)中的硫源(以下,也称为“装料硫源”或“有效硫源”)1.0摩尔优选为0.5~2.4摩尔。在脱水工序中水分量变得过少的情况下,只要在聚合工序之前的装料工序中添加水来调节成期望的水分量即可。
35.(装料工序)
36.装料工序是制备含有有机极性溶剂、硫源、以及多卤芳香族化合物的混合物的工序。也将在装料工序中装入的混合物称为“装料混合物”。
37.在进行脱水工序的情况下,装料混合物中的硫源的量(以下,也称为“装料硫源的量”或“有效硫源的量”)可以通过从作为原料投入的硫源的摩尔量减去脱水工序中挥发的硫化氢的摩尔量来计算出。
38.在进行脱水工序的情况下,在装料工序中,可以根据需要在脱水工序后残存于体系内的混合物中添加碱金属氢氧化物和水。特别是,可以在考虑了脱水时生成的硫化氢的量和脱水时生成的碱金属氢氧化物的量的基础上,添加碱金属氢氧化物。需要说明的是,碱金属氢氧化物的摩尔数基于在装料工序中添加的碱金属氢氧化物的摩尔数、以及在进行脱水工序的情况下在脱水工序中根据需要添加的碱金属氢氧化物的摩尔数、以及在脱水工序中伴随着硫化氢的生成而生成的碱金属氢氧化物的摩尔数来计算出。在硫源包含碱金属硫化物的情况下,每1摩尔硫源(装料硫源)中的碱金属氢氧化物的摩尔数以包含碱金属硫化物的摩尔数的方式计算出。在硫源中使用硫化氢的情况下,以包含所生成的碱金属硫化物的摩尔数的方式计算出每1摩尔硫源(装料硫源)中的碱金属氢氧化物的摩尔数。但是,以其他目的添加的碱金属氢氧化物的摩尔数,例如,在作为聚合助剂和/或相分离剂以有机羧酸与碱金属氢氧化物的组合的形态使用有机羧酸金属盐的情况下,在中和等反应中消耗掉的碱金属氢氧化物的摩尔数不包含于每1摩尔硫源(装料硫源)中的碱金属氢氧化物的摩尔数。而且,在因某些理由而使用选自由无机酸和有机酸构成的组中的至少一种酸的情况下等,中和上述至少一种酸所需的碱金属氢氧化物的摩尔数不包含于每1摩尔硫源(装料硫源)中的碱金属氢氧化物的摩尔数。
39.在装料混合物中,有机极性溶剂和多卤芳香族化合物各自的使用量例如相对于硫源的装料量1摩尔设定在与有机极性溶剂和多卤芳香族化合物相关的上述说明中所示的范围内。
40.(第一聚合工序)
41.第一聚合工序为如下工序:在选自由羧酸盐、碱金属氯化物、有机磺酸盐、硫酸碱金属盐、碱土金属氧化物、碱金属磷酸盐、以及碱土金属磷酸盐构成的组中的至少一种助剂的存在下,对含有有机极性溶剂、硫源、水、多卤芳香族化合物、以及碱金属氢氧化物的混合物进行加热而引发聚合反应,生成含有多卤芳香族化合物的转化率为50摩尔%以上的预聚物的反应混合物。在第一聚合工序中,在生成的聚合物均匀地溶解于有机极性溶剂中的反应体系中进行聚合反应。需要说明的是,在本说明书中,反应混合物是指,包含在上述聚合反应中生成的反应产物的混合物,在开始上述聚合反应的同时,开始生成。
42.为了缩短聚合循环时间的目的,作为聚合反应方式,可以采用使用两个以上反应槽的方式。
43.在第一聚合工序中,优选的是,将装料工序中制备出的混合物,即,将装料混合物加热至温度170℃~270℃的温度,引发聚合反应,生成多卤芳香族化合物的转化率为50摩尔%以上的预聚物。从抑制副反应、分解反应的方面考虑,第一聚合工序中的聚合温度优选选自180~265℃的范围内。
44.多卤芳香族化合物的转化率优选为50~98%,更优选为60~97%,更进一步优选为65~96%,特别优选为70~95%。多卤芳香族化合物的转化率可以通过气相色谱法求出
反应混合物中残存的多卤芳香族化合物的量,基于该残存量、多卤芳香族化合物的装料量以及硫源的装料量计算出。
45.可以在聚合反应的中途,使水和有机极性溶剂的至少一种的量变化。例如,可以在聚合中途将水加入反应体系。其中,在第一聚合工序中通常优选使用装料工序中制备出的装料混合物引发聚合反应,并且结束第一聚合工序中的反应。
46.在第一聚合工序开始时,水的含量相对于每1.0摩尔硫源优选为0.5~2.4摩尔,更优选为0.5~2.0摩尔,更进一步优选为1.0~1.5摩尔。在第一聚合工序开始时,通过将水的含量设为上述范围内,使硫源可溶于有机极性溶剂,能适当地推进反应。
47.第一段聚合工序在选自由羧酸盐、碱金属氯化物、有机磺酸盐、硫酸碱金属盐、碱土金属氧化物、碱金属磷酸盐、以及碱土金属磷酸盐构成的组中的至少一种助剂的存在下进行。由此,在本发明的pas的制造方法中,容易得到高分子量pas。从获取性、操作性等观点考虑,助剂优选羧酸盐。
48.只要在第一段聚合工序中存在助剂,作为添加助剂的阶段就没有特别限定,例如,可列举出:脱水工序、装料工序、或第一聚合工序。
49.助剂可以以羧酸盐等上述的化合物其本身的形式添加到反应体系内,也可以设为以对应的有机酸或无机酸的形态添加到反应体系内,通过与反应体系内的碱金属氢氧化物的中和反应而生成相当于助剂的化合物,由此在第一段聚合工序中存在。
50.助剂的量相对于每1摩尔硫源优选为0.1~50摩尔%,更优选为1~40摩尔%,更进一步优选为5~30摩尔%。若助剂的量在上述范围内,则高分子量pas更容易得到。
51.(相分离剂添加工序)
52.相分离剂添加工序是在第一聚合工序后,向所述反应混合物添加相分离剂的工序。作为相分离剂,没有特别限定,例如,可列举出选自由水、有机羧酸金属盐、有机磺酸金属盐、碱金属卤化物、碱土金属卤化物、芳香族羧酸的碱土金属盐、磷酸碱金属盐、醇类、以及无极性溶剂构成的组中的至少一种。其中,优选成本低廉且后处理容易的水。此外,也优选有机羧酸盐与水的组合,特别是包含碱金属羧酸盐和水的混合物。上述的盐类也可以采用分别添加对应的酸和碱的形态。
53.作为无极性溶剂,例如,可列举出烃,为了促进预聚物彼此的反应,从不溶解预聚物的一方容易得到更高分子量的pas方面考虑,无极性溶剂优选为脂肪族烃,更优选为烷烃(石蜡系烃类),更进一步优选为直链状烷烃。就烃、脂肪族烃、烷烃、以及直链状烷烃的碳原子数而言,只要在第二聚合工序中将烃、脂肪族烃、烷烃、以及直链状烷烃用作溶剂,就没有特别限定,例如,可列举出6~24,从操作性等观点考虑,优选为7~20,更优选为8~18,更进一步优选为9~16,特别优选为10~14。作为无极性溶剂的具体例子,可列举出:正己烷、正庚烷、正辛烷、异辛烷、正壬烷、正癸烷、正十二烷、正十四烷、正十六烷、正十八烷、正二十烷、正二十四烷等,从容易得到更高分子量的pas;操作性、获取性等优异的方面考虑,优选异辛烷、正癸烷、以及正十四烷,更优选正癸烷和正十四烷。
54.相分离剂的使用量根据使用的化合物的种类而不同,但相对于有机极性溶剂1kg,通常在1摩尔~10摩尔的范围内。特别优选的是,采用以第二聚合工序中的反应体系内的水分量相对于每1kg有机极性溶剂为超过4摩尔且20摩尔以下的方式在相分离剂添加工序中添加水作为相分离剂的方法。在本发明中,相分离剂含水,相分离剂添加工序中的水相对于
有机极性溶剂的摩尔比为0.5~3.0,从粒子强度的观点考虑,优选为0.6~2.0,更优选为0.65~1.5。通过使相分离剂的使用量在上述范围,可以高产率制造粒子强度高的pas粒子。
55.需要说明的是,在将包含碱金属羧酸盐和水的混合物用作相分离剂的情况下,该混合物的使用量优选调整为碱金属羧酸盐的量相对于每1摩尔硫源成为30摩尔以下。作为本实施方式的相分离剂的添加方法,没有特别限定,例如可列举:一次添加总量的方法、分多次添加的方法。
56.(第二聚合工序)
57.第二聚合工序是在相分离剂添加工序后,继续聚合反应的工序。在第二聚合工序中,进行如下相分离聚合:在相分离剂的存在下,在反应体系内相分离为富聚合物相和贫聚合物相的状态下继续聚合反应。具体而言,通过添加相分离剂,使聚合反应体系(聚合反应混合物)相分离为富聚合物相(以熔融pas为主的相)和贫聚合物相(以有机极性溶剂为主的相)。
58.针对第二聚合工序中的聚合温度,加热至245℃~290℃,优选加热至250℃~285℃,更优选加热至255℃~280℃来继续聚合反应。聚合温度可以维持为固定的温度,也可以根据需要阶段性地升温或降温。从控制聚合反应的观点考虑,优选维持为固定的温度。聚合反应时间一般为10分钟~72小时的范围,理想的是30分钟~48小时。
59.从产率提高的观点考虑,可以将第二聚合工序后的反应混合物的ph设为8~11,也可以设为9~10.5。作为调整反应混合物的ph的方法,没有特别限定,例如可列举出:调整装料工序中的碱金属氢氧化物的含量的方法;之后添加碱金属氢氧化物、无机酸和/或有机酸的方法。
60.(冷却工序)
61.冷却工序是在第二聚合工序后,对所述反应混合物进行冷却的工序。在冷却工序中,对含有生成聚合物的液相进行冷却。在冷却工序中,在比最大增粘温度高5℃以上且小于250℃的温度下向反应混合物中添加冷却剂,并且最大增粘温度下的冷却速度为2.2℃/分钟以上且6.0℃/分钟以下。
62.通过在上述的范围内的温度下进行向反应混合物添加冷却剂,并且将最大增粘温度下的冷却速度设为上述的范围内,能缩短用于形成粒状pas的冷却工序所花费的时间,并且能抑制所得到的粒状pas的平均粒径增大。
63.冷却速度的下限为2.2℃/分钟以上,优选为2.3℃/分钟以上,更优选为2.4℃/分钟以上,更进一步优选为2.5℃/分钟以上,特别优选为2.6℃/分钟以上。若上述冷却速度为2.2℃/分钟以上,则能缩短用于形成粒状pas的冷却工序所花费的时间,并且能抑制所得到的粒状pas的平均粒径增大。上述冷却速度的上限没有特别限定,例如,可以为6.0℃/分钟以下,也可以为5.0℃/分钟以下、4.0℃/分钟以下、3.9℃/分钟以下。
64.在冷却工序中,只要冷却剂向反应混合物的添加在上述的规定的范围内的温度下进行,并且最大增粘温度下的冷却速度在上述的范围内,冷却速度的分布就没有特别限定。由此,最大增粘温度以外的温度下的冷却速度例如可以是利用自然空冷的冷却速度,从缩短冷却工序的时间的观点考虑,也可以与最大增粘温度下的冷却速度相同。此外,例如,在使用后述的冷却剂进行冷却的情况下,由于反应釜的容量有限而因此冷却剂的添加量受限制时,也可以首先进行自然空冷,然后,在反应釜的内容物的温度降低至最大增粘温度的附
近之后,进行将超过最大增粘温度的温度下的冷却速度设为与最大增粘温度下的冷却速度相同的冷却速度调节。这时,作为进行上述冷却速度调节的温度,只要根据反应釜的容量等适当设定即可,例如可列举出超过最大增粘温度(最大增粘温度+20℃)以下,也可以为(最大增粘温度+5℃)以上且(最大增粘温度+15℃)以下,也可以为(最大增粘温度+8℃)以上且(最大增粘温度+12℃)以下,也可以为最大增粘温度+10℃。
65.在冷却工序中,在比最大增粘温度高5℃以上且小于250℃的温度下向反应混合物添加冷却剂。因此,最大增粘温度下的冷却速度为2.2℃/分钟以上且6.0℃/分钟以下这样的冷却方法至少包括向反应混合物添加冷却剂。也可以将基于添加冷却剂的冷却与其他冷却方法组合实施。其他冷却方法没有特别限定,例如可列举出:利用风扇、循环器等气流产生装置的强制空冷;聚合反应槽的夹套中的制冷剂的循环;利用回流冷凝器的、反应混合物中的气相的回流等。其中,从容易防止由聚合反应槽的壁面的冷却而导致pas向该壁面附着的方面等考虑,优选向反应混合物添加冷却剂。作为冷却剂,没有特别限定,从在后处理工序中容易从pas分离的方面考虑,优选水和/或冰;有机极性溶剂(例如,nmp等有机酰胺溶剂),从比热、汽化潜热(latent heat of vaporization)大的方面考虑,更优选水和/或冰。此外,从反应混合物的相分离性容易提高、pas的产率容易提高的方面考虑,作为冷却剂,也更优选水和/或冰。
66.在最大增粘温度下,从根据量的充分的冷却容易进行的方面考虑,反应混合物中的冷却剂的含量相对于所述硫源1摩尔优选为2.7摩尔以上且6.0摩尔以下,更优选为3.0摩尔以上且5.5摩尔以下,更进一步优选为4.0摩尔以上且5.0摩尔以下。
67.在本说明书中,最大增粘温度是指,通过自然空冷对所述第二聚合工序后的所述反应混合物从255℃开始进行冷却时,在240℃到220℃之间测定该反应混合物的搅拌转矩,该搅拌转矩为最大的温度。
68.(后处理工序(分离工序、清洗工序、回收工序等))
69.在本实施方式的pas的制造方法中,可以通过常规方法、例如通过日本特开2016-056232号公报所记载的方法来进行聚合反应后的后处理工序。
70.(所得到的pas)
71.就通过本实施方式中的pas的制造方法得到的pas而言,平均粒径优选为2000μm以下,更优选为1800μm以下,更进一步优选为1500μm以下。上述pas的平均粒径的下限没有特别限定,例如,可以为200μm以上,也可以为300μm以上、400μm以上。需要说明的是,在本说明书中,平均粒径是指通过筛分而如后述的实施例所记载那样测定出的值。
72.就通过本实施方式中的pas的制造方法得到的pas而言,在温度310℃和剪切速度1216sec-1
下测定出的熔融粘度优选为1000pa
·
s以下,更优选为300pa
·
s以下,更进一步优选为150pa
·
s以下。上述pas的上述熔融粘度的下限没有特别限定,例如可以为1pa
·
s以上,也可以为5pa
·
s以上、8pa
·
s以上。需要说明的是,在本说明书中,熔融粘度是指,使用pas的干燥聚合物约20g并使用毛细管流变仪(capilograph)在上述温度和剪切速度下测定出的值。
73.就通过本实施方式中的pas的制造方法得到的pas而言,可以在直接或使其氧化交联后,单独或根据期望配合各种无机填充剂、纤维状填充剂、各种合成树脂,成型为各种注塑成型品或片、膜、纤维、以及管等挤出成型品。
74.在本实施方式的pas的制造方法中,pas不特别限定,优选为pps。
75.本发明并不限定于上述的实施方式,可以在权利要求所示的范围内进行各种变更,对分别公开的技术方案进行适当组合而得到的实施方式也包含在本发明的技术范围内。此外,本说明书中记载的文献全部作为参考被引用。
76.实施例
77.以下,列举实施例和比较例,对本发明更具体地进行说明。需要说明的是,本发明并不限于实施例。实施例和比较例中的操作只要没有特别说明就在室温下进行。此外,在本说明书中,重均分子量(以下,也称为“mw”)是指由凝胶渗透色谱法测定出的聚苯乙烯换算的重均分子量。
78.(1)熔融粘度
79.pas的熔融粘度通过作为毛细管的装接有长度10.0mm的喷嘴的(株)东洋精机制作所制capilograph(注册商标)1d进行了测定。将设定温度设为310℃。将聚合物试样导入装置内,保持5分钟后,在剪切速度1200sec-1
下测定了熔融粘度。
80.(2)平均粒径
81.就pas的平均粒径而言,通过作为使用筛网而使用筛网孔2800μm(7目(网孔数/英寸))、筛网孔1410μm(12目(网孔数/英寸))、筛网孔1000μm(16目(网孔数/英寸))、筛网孔710μm(24目(网孔数/英寸))、筛网孔500μm(32目(网孔数/英寸))、筛网孔250μm(60目(网孔数/英寸))、筛网孔150μm(100目(网孔数/英寸))、筛网孔105μm(145目(网孔数/英寸))、筛网孔75μm(200目(网孔数/英寸))、筛网孔38μm(400目(网孔数/英寸))的筛网的筛分法进行测定,根据各筛网的筛上物的质量,计算出累积质量为50%质量时的平均粒径。
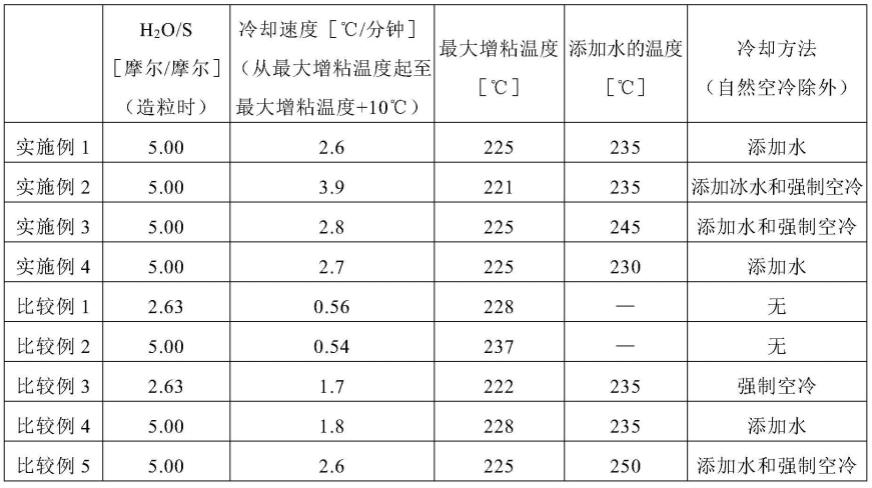
82.[实施例1]
[0083]
(脱水工序)
[0084]
向20升的高压釜中装入6005g的nmp、2003g的氢硫化钠水溶液(nash:纯度62.24质量%)、1071g的氢氧化钠(naoh:纯度73.40质量%)、以及180g的乙酸钠。将该高压釜内用氮气置换后,历时约4小时,一边通过搅拌机以转速250rpm进行搅拌一边缓慢升温至200℃,馏出了902g的水(h2o)、763g的nmp、以及15g的硫化氢(h2s)。
[0085]
(第一聚合工序)
[0086]
上述脱水工序后,将高压釜的内容物冷却至150℃,加入3244g的pdcb、3302g的nmp、8g的氢氧化钠、以及107g的水,一边搅拌一边升温至220℃后,用1.5小时升温至260℃,进行第一聚合工序。釜内的nmp/装料硫源(以下,简记为“装料s”)的比率(g/摩尔)为391,pdcb/装料s(摩尔/摩尔)为1.010,h2o/装料s(摩尔/摩尔)为1.50。第一聚合工序中的pdcb的转化率为93%。
[0087]
(相分离剂添加工序)
[0088]
第一聚合工序结束后,一边搅拌高压釜的内容物一边压入60g的氢氧化钠和445g的离子交换水,将搅拌机的转速提高到400rpm。h2o/nmp(摩尔/摩尔)为0.67,h2o/装料s(摩尔/摩尔)为2.63。
[0089]
(第二聚合工序)
[0090]
压入离子交换水后,将高压釜的内容物升温至265℃,使其反应2.5小时进行第二聚合工序。
[0091]
(冷却工序)
[0092]
第二聚合工序结束后,通过自然空冷以冷却速度0.56℃/分钟对高压釜的内容物从265℃开始进行冷却,在所述内容物的温度成为最大增粘温度+10℃的时间点,以h2o/装料s(摩尔/摩尔)从2.63变为5.00的方式向高压釜内压入室温的水,使冷却速度上升,然后,进行冷却至室温。需要说明的是,最大增粘温度下的冷却速度为2.6℃/分钟。
[0093]
需要说明的是,最大增粘温度的测定方法如下所述。已知,在能制造高分子量pas的、伴随着相分离聚合系的、聚合反应结束后的反应釜内,pas以熔融状态存在,通过相分离剂而相分离为富相和贫相。而且,若在搅拌下对体系进行冷却,则pas从熔融状态固体化,成为粉状或粒子状或颗粒状的固体,以悬浮的状态存在,在该过程中发生体系的粘度变化。即,若在搅拌下对包含熔融的pas的分散系进行冷却,则由于pas伴随着冷却的增粘而体系整体的表观粘度逐渐增大,当达到某个温度时,体系整体的表观粘度反而会降低。该体系的最大增粘温度可以以一定搅拌下的搅拌转矩或者向搅拌机的供给动力的形式检测出。最大增粘温度基于电力消耗的极大值而预先测定出,所述电力消耗的极大值是将钳式电力计cw140(横河电机株式会社)设置于高压釜的搅拌机马达而测定出的。
[0094]
(后处理工序)
[0095]
冷却工序后,用网孔径150μm(100目)的筛网对高压釜的内容物进行筛分,用丙酮和离子交换水进行清洗后,用乙酸水溶液进行清洗,进行一昼夜干燥,得到了粒状pps。将冷却工序中的条件等示于表1,将粒状聚合物的物性等示于表2(以下相同)。
[0096]
[实施例2]
[0097]
在冷却工序中,向高压釜内压入冰水来代替室温的水,而且,在压入冰水的同时,开始利用风扇的高压釜的强制空冷,除此以外,与实施例1同样地得到了粒状pps。需要说明的是,最大增粘温度下的冷却速度为3.9℃/分钟。
[0098]
[实施例3]
[0099]
将压入室温的水的温度变为最大增粘温度+20℃,除此以外,与实施例1同样地得到了粒状pps。需要说明的是,最大增粘温度下的冷却速度为2.8℃/分钟。
[0100]
[实施例4]
[0101]
将压入室温的水的温度变为最大增粘温度+5℃,除此以外,与实施例1同样地得到了粒状pps。需要说明的是,最大增粘温度下的冷却速度为2.7℃/分钟。
[0102]
[比较例1]
[0103]
在冷却工序中,通过自然空冷以冷却速度0.56℃/分钟将高压釜的内容物从265℃开始冷却至室温,除此以外,与实施例1同样地得到了粒状pps。需要说明的是,最大增粘温度下的冷却速度为0.56℃/分钟。
[0104]
[比较例2]
[0105]
为了确认由冷却工序中的水的含量增加带来的影响,在冷却工序中,在第二聚合工序结束后立即向高压釜内压入室温的水,使h2o/装料s(摩尔/摩尔)从2.63变为5.00,此外,最大增粘温度下的冷却速度为接近比较例1中的值的0.54℃/分钟,除此以外,与实施例1同样地得到了粒状pps。
[0106]
[比较例3]
[0107]
为了确认由冷却速度带来的影响,在冷却工序中,进行利用风扇的高压釜的强制
空冷来代替以h2o/装料s(摩尔/摩尔)从2.63变为5.00的方式向高压釜内压入室温的水,此外,最大增粘温度下的冷却速度为1.7℃/分钟,除此以外,与实施例1同样地得到了粒状pps。
[0108]
[比较例4]
[0109]
为了确认用于抑制粒状pas的平均粒径增大所需的冷却速度,在冷却工序中调整水的压入速度,其结果是,最大增粘温度下的冷却速度为1.8℃/分钟,除此以外,与实施例1同样地得到了粒状pps。
[0110]
[比较例5]
[0111]
为了确认用于抑制粒状pas的平均粒径增大所需的冷却剂的添加温度,在冷却工序中将添加水的温度设为250℃,并且在添加水后进行利用风扇的高压釜的强制空冷,除此以外,与实施例1同样地得到了粒状pps。
[0112]
[表1]
[0113][0114]
[表2]
[0115]
[0116][0117]
如表1和表2所明确的那样,根据本发明的pas的制造方法,能缩短用于形成粒状pas的冷却工序所花费的时间,并且,能抑制所得到的粒状pas的平均粒径增大。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1