含环碳酸酯基的(甲基)丙烯酸酯单体及聚合物的制作方法
1.本发明涉及一种含有环碳酸酯基的(甲基)丙烯酸酯单体及含有该单体的聚合物,该聚合物能够抑制经时性增稠,可长期稳定地用于涂料等用途。
背景技术:
2.环碳酸酯基通常具有高极性、高介电常数、且对高分子的溶解性高等特征,由于其特征性结构,因此能够应用于各种用途。特别是在为同时具有环碳酸酯基与(甲基)丙烯酰基的化合物的情况下,通过与其他单体、低聚物类进行聚合,能够得到导入了环碳酸酯基的聚合物。使用这样的含环碳酸酯基的(甲基)丙烯酸酯单体而得到的聚合物,例如能够用作膜
·
成型材料、密封剂、涂料、粘合剂、各种粘结剂等。
3.含环碳酸酯基的(甲基)丙烯酸酯单体通常能够通过使二氧化碳对环氧化合物进行作用而得到,专利文献1、2中,对以高收率得到该单体的方法进行了报道。这些现有文献中列举出的含环碳酸酯基的(甲基)丙烯酸酯单体能够用于合成聚合物,可得到在侧链上具有环碳酸酯基的聚合物。这样的聚合物例如如专利文献3报道所述,透明性、粘度等各种物性优异,能够毫无问题地使用。现有技术文献专利文献
4.专利文献1:日本特开平第5-202022专利文献2:日本特开第2011-32222专利文献3:日本特开第2014-105265
技术实现要素:
本发明要解决的技术问题
5.近年来,特别是在涂料及油墨用途中,伴随着高画质化及高速化的印刷技术的提高、对复杂形状对象物的应对或多种色彩的混合的高设计性的要求,需要极其精密地控制涂料或油墨的粘度。然而,在这些用途中,若使用通过专利文献1~3中记载的以往方法制备的在侧链上具有环碳酸酯基的聚合物,则在严酷环境下使用时或经过长期保管时粘度有时会稍微上升,视情况还可能会在印刷时产生模糊。
6.因此,要求以下特性,即要求即使是使用了能够以高收率得到的含环碳酸酯基的(甲基)丙烯酸酯单体的聚合物,也不会发生在印刷时产生模糊等的不良情况,且经时性粘度变化小。
7.本发明的技术问题在于提供一种含环碳酸酯基的(甲基)丙烯酸酯单体及含有该单体的聚合物,其中,在将含环碳酸酯基的(甲基)丙烯酸酯单体制成聚合物而用于涂料等时,经时性增稠得以抑制,能够稳定地长期使用。解决技术问题的技术手段
8.本技术的发明人为了解决上述技术问题进行了认真研究,结果发现,在以往已知
的关于含环碳酸酯基的(甲基)丙烯酸酯单体的报告中,虽然有时提及了作为该单体原料的各种具有(甲基)丙烯酰基的环氧化合物,但对于以一定量而含有的作为副产物的二(甲基)丙烯酸酯单体的含量未进行研究。
9.因此,着眼于二(甲基)丙烯酸酯单体的含量进行了研究,结果发现,在进行聚合而制成聚合物并用于涂料等用途时,经时性增稠得以抑制,能够长期稳定地用于涂料等用途。
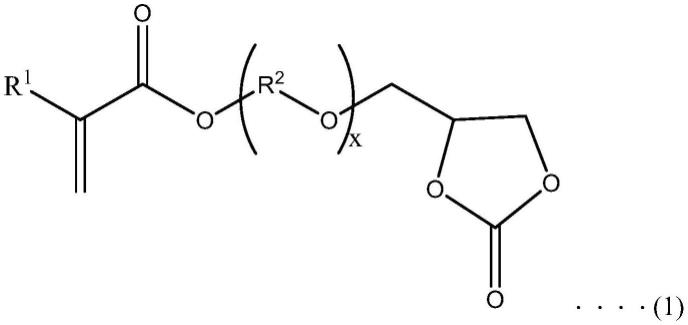
10.即,本发明涉及下述(1)及(2)。(1)一种(a)下述式(1)所表示的含环碳酸酯基的(甲基)丙烯酸酯单体,其特征在于,(b)下述式(2)所表示的二(甲基)丙烯酸酯单体的含量为3重量%以下。[化学式1]式(1)中,r1表示氢原子或甲基,r2表示碳原子数为2~10的烷基,x=0或1。[化学式2]式(2)中,r3表示氢原子或甲基,r4表示碳原子数为2~10的烷基,x=0或1。
[0011]
(2)一种聚合物,其特征在于,(1)的含环碳酸酯基的(甲基)丙烯酸酯单体(a)的质量比为1~100质量%,能够与所述单体(a)共聚的其他单体(c)的质量比为0~99质量%。发明效果
[0012]
本发明的化合物为一种含环碳酸酯基的(甲基)丙烯酸酯单体,其特征在于,二(甲基)丙烯酸酯单体的含量少于以往。在将该含环碳酸酯基的(甲基)丙烯酸酯单体作为单体,或进行聚合而制成聚合物并用于涂料等时,经时性增稠得以抑制,能够长期稳定地用于涂料等用途。
具体实施方式
[0013]
以下,进一步对本发明的实施方式进行详细说明。[(a)含环碳酸酯基的(甲基)丙烯酸酯单体]本发明的含环碳酸酯基的(甲基)丙烯酸酯单体(a)由下述式(1)所表示。
[化学式1]
[0014]
本发明的含环碳酸酯基的(甲基)丙烯酸酯单体(a)的特征在于,在一分子中具有1个环碳酸酯基和1个(甲基)丙烯酰基。
[0015]
在通式(1)中,r2表示碳原子数为2~10的烷基。通过使r2为碳原子数为2~10的烷基,合成聚合物时的反应性良好,能够得到单体残留成分少的聚合物。其中,r2的碳原子数优选为2~4,r2的碳原子数特别优选为2。
[0016]
式(1)中,x的值为0或1。其中,优选x=0。通过使x为0或1,合成聚合物时的反应性良好,能够得到单体残留成分少的聚合物。
[0017]
[二(甲基)丙烯酸酯单体(b)]二(甲基)丙烯酸酯单体(b)由下述式(2)所表示。[化学式2]式(2)中,r3表示氢原子或甲基,r4表示碳原子数为2~10的烷基,x=0或1。
[0018]
二(甲基)丙烯酸酯单体(b)作为副产物而包含在含环碳酸酯基的(甲基)丙烯酸酯单体(a)中。另外,二(甲基)丙烯酸酯单体(b)的结构根据选择何种单体作为起始原料的单体而变化。例如,在以(甲基)丙烯酸缩水甘油酯为原料时,产生丙三醇二(甲基)丙烯酸酯。通过上述方式生成的二(甲基)丙烯酸酯单体(b)的结构及其含量可通过气相色谱法或高速液体色谱法、核磁共振等适当的方法而确定。
[0019]
由于伴随含环碳酸酯基的(甲基)丙烯酸酯单体(a)的制备,二(甲基)丙烯酸酯单体(b)作为副产物而生成,因此r4表示碳原子数为2~10的烷基。其中,r4的碳原子数优选为2~4,特别优选为2。
[0020]
式(2)中,x的值为0或1。其中,优选x=0。通过使x为该值,合成聚合物时的反应性良好,能够得到单体残留成分少的聚合物。
[0021]
(二(甲基)丙烯酸酯单体(b)的含有率)在将式(1)所表示的含环碳酸酯基的(甲基)丙烯酸酯单体(a)的量设为100质量%
时,将单体(a)中所含有的式(2)所表示的二(甲基)丙烯酸酯单体(b)的含量设为3质量%以下,进一步优选设为1.5质量%以下,最优选设为1.0质量%以下。若含有多于3质量%的该单体,则在使用本发明的含环碳酸酯基的(甲基)丙烯酸酯单体合成聚合物时,在保管时发生高分子量化、粘度上升、凝胶化反应。
[0022]
此外,在将含环碳酸酯基的(甲基)丙烯酸酯单体(a)的量设为100质量%时,单体(a)中所含有的二(甲基)丙烯酸酯单体(b)的含量的下限没有特别限定,可以为0质量%。但从抑制经时性增稠与固化膜物性的角度出发,优选将单体(a)中所含有的二(甲基)丙烯酸酯单体(b)的含量设为0.1质量%以上,进一步优选设为0.3质量%以上,特别优选设为0.5质量%以上。
[0023]
[含环碳酸酯基的(甲基)丙烯酸酯单体的制备方法]含环碳酸酯基的(甲基)丙烯酸酯单体(a)可通过以下方式得到:以各种具有(甲基)丙烯酰基的环氧化合物为起始物质,以0.05~0.3mpa左右吹入二氧化碳使其反应。此时,作为具有(甲基)丙烯酰基的环氧化合物,可使用各种单体。然而,由于粘度低,反应所需的时间短,所得到的含环碳酸酯基的(甲基)丙烯酸酯单体(a)的色相低,可得到透明性良好的单体,因此优选使用(甲基)丙烯酸缩水甘油酯、4-羟基丁基(甲基)丙烯酸酯缩水甘油醚,进一步优选使用(甲基)丙烯酸缩水甘油酯。
[0024]
在本发明中,使二氧化碳对环氧基进行作用,制成环碳酸酯基。作为向反应系中导入二氧化碳的方法,可选择在加压下的各种吹入方法。此时,通过将压力、温度控制在一定的范围内,能够提高目标产物的收率,将副产物的含有率抑制得较低。此时的压力为0.05~0.3mpag,优选为0.1~0.2mpag。即使将压力设定得较高使其超过0.3mpag,环碳酸酯体的收率也不会上升,容易进行副反应,因此作为副产物而生成的二(甲基)丙烯酸酯单体(b)的含量变得大于3质量%。
[0025]
合成时的反应温度虽根据压力或催化的条件而不同,但其为40~70℃的范围,优选50~60℃的范围。若小于40℃,则反应所需的时间变得过长,并且容易残留未反应的原料。此外,容易发生着色。另一方面,若超过70℃,则导致大量含有二(甲基)丙烯酸酯单体(b),在聚合物化时,也容易引起凝胶化等不良情况。此外,容易发生着色。
[0026]
[(c)其他单体)]本发明的聚合物可通过使上述单体(b)的质量为3质量%以下的单体(a)进行聚合而得到。然而,本发明的聚合物也可含有其他单体(c)。单体(c)只要为能够与单体(a)共聚的单体则没有特别限定,可含有一种或两种以上,优选(甲基)丙烯酸酯单体、芳香族乙烯基化合物。
[0027]
作为(甲基)丙烯酸酯单体,例如可列举出(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸戊酯、(甲基)丙烯酸己酯、(甲基)丙烯酸环己酯、(甲基)丙烯酸庚酯、(甲基)丙烯酸辛酯、(甲基)丙烯酸2-乙基己酯、(甲基)丙烯酸壬酯、(甲基)丙烯酸癸酯、(甲基)丙烯酸十二烷酯、(甲基)丙烯酸苯酯、(甲基)丙烯酸苄酯、(甲基)丙烯酸2-甲氧基乙酯、(甲基)丙烯酸3-甲氧基丁酯、(甲基)丙烯酸2-羟基乙酯、(甲基)丙烯酸甘油酯、(甲基)丙烯酸羟基丙酯、(甲基)丙烯酸羟基丁酯、(甲基)丙烯酸硬脂酸酯、(甲基)丙烯酸的环氧乙烷加成物等,优选(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸环己酯、(甲基)丙烯酸2-乙基己
酯、(甲基)丙烯酸苄酯,更优选(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸环己酯、(甲基)丙烯酸2-乙基己酯。
[0028]
作为芳香族乙烯基化合物,可列举出苯乙烯、α-甲基苯乙烯、对甲基苯乙烯、间甲基苯乙烯、邻甲基苯乙烯、对乙基苯乙烯、间乙基苯乙烯、邻乙基苯乙烯、叔丁基苯乙烯、氯苯乙烯、羟基苯乙烯、叔丁氧基苯乙烯、乙烯基甲苯、乙烯基萘等,优选苯乙烯。
[0029]
本发明的聚合物可以为使上述单体(b)的质量为3质量%以下的单体(a)进行聚合而成的聚合物。或者,本发明的聚合物可以为使上述单体(b)的质量为3质量%以下的单体(a)与其他单体(c)进行共聚而成的聚合物。
[0030]
在适宜的实施方式中,在将聚合前的单体的合计质量设为100质量%时,来自单体(a)的结构单元占构成聚合物的结构单元的共聚比例为1~100质量%,优选为5~80质量%,更优选为10~60质量%,特别优选为15~40质量%。
[0031]
此外,在将聚合前的单体的合计质量设为100质量%时,来自其他单体(c)的结构单元占构成本发明的聚合物的结构单元的共聚比例为0~99质量%,优选为20~95质量%,更优选为40~90质量%,特别优选为60~85质量%。
[0032]
[聚合物]本发明的聚合物的重均分子量可使用凝胶渗透色谱法(gpc)以聚苯乙烯换算而求出,优选为3,000~1,000,000,进一步优选为10,000~800,000,更优选为50,000~300,000。若聚合物的重均分子量过小,则在制成涂料时可能会引起膜强度的下降,若重均分子量过高,则溶剂溶解性或溶液粘度变得过高,因而可能会发生操作性的下降。
[0033]
具体而言,在使用本发明的含环碳酸酯基的(甲基)丙烯酸酯单体合成聚合物时,聚合物以40℃保管1个月后的粘度(d)相对于刚合成后的粘度(c)的比率(d/c)为1.00以上,优选为1.20以下,进一步优选为1.15以下,特别优选为1.05以下,最优选为1.02以下。
[0034]
[聚合物的制备方法]接着,对制备本发明的聚合物的方法进行说明。本发明的聚合物可通过使至少含有可包含单体(b)的单体(a),根据需要而进一步含有单体(c)的单体混合物进行自由基聚合而获得。聚合可通过公知的方法进行。例如可列举出溶液聚合、悬浮聚合、乳液聚合等,从容易将共聚物的重均分子量调节在上述范围内的方面出发,优选溶液聚合或悬浮聚合。
[0035]
聚合引发剂可使用公知的引发剂。例如可列举出1,1,3,3-四甲基丁基过氧化-2-乙基己酸酯等有机过氧化物、2,2
’‑
偶氮二异丁腈等偶氮类聚合引发剂等。这些聚合引发剂可仅使用一种,也可同时使用两种以上。
[0036]
聚合引发剂的使用量可根据所使用单体的组合或反应条件等而适当设定。另外,在加入聚合引发剂时,例如可以将全部量一次加入,也可以加入一部分并滴加剩余部分,或者也可以滴加全部量。此外,若同时滴加所述单体与聚合引发剂,则容易控制反应,故而优选,进一步,若在滴加单体后也添加聚合引发剂,则能够减少残留单体,故而优选。
[0037]
作为在溶液聚合时所使用的聚合溶剂,可使用溶解单体与聚合引发剂的溶剂,具体而言,可列举出甲醇、乙醇、1-丙醇、丙酮、甲基乙基酮、丙二醇单甲醚、n,n-二甲基甲酰胺等。
[0038]
单体(合计量)相对于聚合溶剂的浓度优选为10~60质量%,特别优选为20~50质量%。若单体混合物的浓度过低,则单体容易残留,有可能所得到的共聚物的分子量会下降,若单体的浓度过高,则可能难以控制发热。
[0039]
在加入单体时,例如可以将全部量一次加入,也可以加入一部分并滴加剩余部分,或者也可以滴加全部量。从控制发热的容易程度出发,优选加入一部分并滴加剩余部分、或滴加全部量。
[0040]
聚合温度依赖于聚合溶剂的种类等,例如为50℃~110℃。聚合时间依赖于聚合引发剂的种类与聚合温度,例如在将双(4-叔丁基环己基)过氧化二碳酸酯用作聚合引发剂时,若将聚合温度设为70℃而进行聚合,则聚合时间适宜为6小时左右。通过进行以上的聚合反应,可得到本发明的聚合物。所得到的聚合物可直接使用,也可对聚合反应后的反应液实施滤取或提纯而进行分离。实施例
[0041]
以下列举实施例及比较例对本发明进行具体说明。
[0042]
(实验1)[评价方法](成分定量分析)使用气相色谱法(gc),根据以下的条件进行含环碳酸酯基的(甲基)丙烯酸酯单体(a)、二(甲基)丙烯酸酯单体(b)的成分定量分析。根据(a)的峰面积与(b)的峰面积的面积比,计算含环碳酸酯基的(甲基)丙烯酸酯单体(a)、二(甲基)丙烯酸酯单体(b)的各收率。将(a)的收率作为(a)的含量(质量%),将(b)的收率作为(b)的含量(质量%)。(单体(a)中所含有的单体(b)的质量比率(质量%)=单体(b)的收率(含量:质量%)/(单体(a)的收率(含量:质量%)+单体(b)的收率(含量:质量%))=单体(b)的峰面积/(单体(a)的峰面积+单体(b)的峰面积)此外,通过下述式计算转化率。转化率(%)=[(含环碳酸酯基的(甲基)丙烯酸酯单体(a)的面积)/(全部峰的面积的总计)]
×
100
[0043]
(gc的条件)装置:gc-2014(shimadzu corporation制造)色谱柱:db-1注入温度:200℃检测器温度:250℃加热简况(heating profile):以40℃保持10分钟
→
以10℃/分钟进行升温
→
升温至250℃并保持注入量:1μl检测器:fid range1载气:氦气70kpa分流比:1/50定量方法:内标法(使用联苯)
[0044]
(聚合物化试验)
使用所得到的聚合性组合物,以下述条件进行聚合物化。掺合组分:聚合性组合物50g甲基丙烯酸甲酯50g引发剂:2,2
’‑
偶氮双(2,4-二甲基戊腈)(产品名称:v-65(wako pure chemical industries,ltd.制造))0.4g溶剂:异丙醇150g反应温度:75℃反应时间:3小时
[0045]
(粘度评价)对所得到的聚合物溶液进行粘度评价。具体而言,测定刚合成聚合物溶液后的粘度(c)、及在40℃的恒温槽中保管1个月后的粘度(d)。以“保管后的粘度/刚合成后的粘度(d/c)”表示两个粘度的比,通过以下基准进行评价。
○
:粘度比(d/c)为1.0以上且小于1.1
△
:粘度比(d/c)为1.1以上且小于1.3
×
:粘度比(d/c)为1.3以上或无法测定
[0046]
按照以下方式合成各例的含环碳酸酯基的(甲基)丙烯酸酯单体。《实施例1-1》向具备二氧化碳导入管、搅拌机、温度计的高压釜中加入1,000份作为起始原料的blemmer gh(甲基丙烯酸缩水甘油酯)、50份碘化钠、0.5份甲氧基对苯二酚。边将系内温度调节至50℃,边通过二氧化碳气瓶将系内保持在0.2mpa并间歇地持续吹入二氧化碳,边搅拌8小时边使其反应。冷却至室温后,加入300份离子交换水并充分搅拌,暂时静置,使有机相与水相分液,去除水相。重复该操作4次,去除碘化钠。然后,在70℃、减压下对有机相进行2小时脱水,得到目标单体。
[0047]
《实施例1-2》向具备二氧化碳导入管、搅拌机、温度计的高压釜中加入1,000份blemmer gh(甲基丙烯酸缩水甘油酯)、50份溴化锂、53.5份二氮杂二环十一碳烯、0.5份甲氧基对苯二酚。边将系内温度调节至60℃,边通过二氧化碳气瓶将系内保持在0.05mpa并间歇地持续吹入二氧化碳,边搅拌8小时边使其反应。冷却至室温后,加入300份离子交换水并充分搅拌,暂时静置,使有机相与水相分液,去除水相。重复该操作4次,去除碘化钠。然后,在70℃、减压下对有机相进行2小时脱水,得到目标单体。
[0048]
《实施例1-3》向具备二氧化碳导入管、搅拌机、温度计的高压釜中加入1,000份blemmer gh(甲基丙烯酸缩水甘油酯)、50份溴化锂、53.5份二氮杂二环十一碳烯、0.5份甲氧基对苯二酚、500份二甲基甲酰胺。以开放排气阀的状态,边将系内温度调节至50℃,边通过二氧化碳气瓶将系内保持在0.05mpa并间歇地持续吹入二氧化碳,边搅拌8小时边使其反应。冷却至室温后,加入300份离子交换水并充分搅拌,暂时静置,使有机相与水相分液,去除水相。重复该操作4次,去除碘化钠。然后,在70℃、减压下对有机相进行2小时脱水,得到目标单体。
[0049]
《实施例1-4》向具备二氧化碳导入管、搅拌机、温度计的高压釜中加入1,000份4-羟基丁基丙烯
酸酯缩水甘油醚、50份碘化钠、0.5份甲氧基对苯二酚。边将系内温度调节至50℃,边通过二氧化碳气瓶将系内保持在0.08mpa并间歇地持续吹入二氧化碳,边搅拌8小时边使其反应。冷却至室温后,加入300份离子交换水并充分搅拌,暂时静置,使有机相与水相分液,去除水相。重复该操作4次,去除碘化钠。然后,在70℃、减压下对有机相进行2小时脱水,得到目标单体。
[0050]
《实施例1-5》向具备二氧化碳导入管、搅拌机、温度计的高压釜中加入1,000份4-羟基丁基丙烯酸酯缩水甘油醚、50份碘化钠、0.5份甲氧基对苯二酚。边将系内温度调节至50℃,边通过二氧化碳气瓶将系内保持在0.2mpa并间歇地持续吹入二氧化碳,边搅拌8小时边使其反应。冷却至室温后,加入300份离子交换水并充分搅拌,暂时静置,使有机相与水相分液,去除水相。重复该操作4次,去除碘化钠。然后,在70℃、减压下对有机相进行2小时脱水,得到目标单体。
[0051]
《比较例1-1》向具备二氧化碳导入管、搅拌机、温度计的高压釜中加入1,000份blemmer gh(甲基丙烯酸缩水甘油酯)、50份碘化钠、0.5份甲氧基对苯二酚。边将系内温度调节至75℃,边通过填充有二氧化碳气体的气球间歇地持续吹入二氧化碳,边搅拌15小时边使其反应。冷却至室温后,加入300份离子交换水并充分搅拌,暂时静置,使有机相与水相分液,去除水相。重复该操作4次,去除碘化钠。然后,在70℃、减压下对有机相进行2小时脱水,得到目标单体。
[0052]
[表1]
[0053]
根据表1所示的结果可知,本发明的实施例1-1~1-5得到了任何物性均优异的单体。
[0054]
另一方面,在比较例1-1中,由于二甲基丙烯酸酯的含量超过了本发明的范围,因此刚合成后的聚合物的粘度较高,进一步,由于在长期保管时发生凝胶化,因此未能测定粘度比。
[0055]
(实验2)
[单体(a)的分析](成分定量分析)使用气相色谱法(gc),通过以下的条件进行含环碳酸酯基的(甲基)丙烯酸酯单体(a)、二(甲基)丙烯酸酯单体(b)的成分定量分析。根据(a)的峰面积与(b)的峰面积的面积比,计算含环碳酸酯基的(甲基)丙烯酸酯单体(a)、二(甲基)丙烯酸酯单体(b)的含量。gc的条件装置:gc-2014(shimadzu corporation制造)色谱柱:db-1注入温度:200℃检测器温度:250℃加热简况:以40℃保持10分钟
→
以10℃/分钟进行升温
→
升温至250℃并保持注入量:1μl检测器:fid range1载气:氦气70kpa分流比:1/50
[0056]
[合成例]通过以下方式合成各例的含环碳酸酯基的(甲基)丙烯酸酯单体。(单体a1的合成)向具备二氧化碳导入管、搅拌机、温度计的高压釜中加入1,000份blemmer gh(甲基丙烯酸缩水甘油酯)、50份溴化锂、53.5份二氮杂二环十一碳烯、0.5份甲氧基对苯二酚、500份二甲基甲酰胺。以开放排气阀的状态,边将系内温度调节至50℃,边通过二氧化碳气瓶将系内保持在0.05mpa并间歇地持续吹入二氧化碳,边搅拌8小时边使其反应。冷却至室温后,加入300份离子交换水并充分搅拌,暂时静置,使有机相与水相分液,去除水相。重复该操作4次,去除碘化钠。然后,在70℃、减压下对有机相进行2小时脱水,得到目标单体a1。所得到的单体a1的纯度为99.2质量%,二(甲基)丙烯酸酯单体量为0.8质量%。
[0057]
(单体a2的合成)向具备二氧化碳导入管、搅拌机、温度计的高压釜中加入1,000份4-羟基丁基丙烯酸酯缩水甘油醚、50份碘化钠、0.5份甲氧基对苯二酚。边将系内温度调节至50℃,边通过二氧化碳气瓶将系内保持在0.08mpa并间歇地持续吹入二氧化碳,边搅拌8小时边使其反应。冷却至室温后,加入300份离子交换水并充分搅拌,暂时静置,使有机相与水相分液,去除水相。重复该操作4次,去除碘化钠。然后,在70℃、减压下对有机相进行2小时脱水,得到目标单体a2。所得到的单体a2的纯度为99.6质量%,二(甲基)丙烯酸酯单体量为0.4质量%。
[0058]
(单体a3的合成))向具备二氧化碳导入管、搅拌机、温度计的高压釜中加入1,000份blemmer gh(甲基丙烯酸缩水甘油酯)、50份碘化钠、0.5份甲氧基对苯二酚。边将系内温度调节至90℃,边通过填充有二氧化碳气体的气球间歇地持续吹入二氧化碳,边搅拌8小时边使其反应。冷却至室温后,加入300份离子交换水并充分搅拌,暂时静置,使有机相与水相分液,去除水相。重复该操作4次,去除碘化钠。然后,在70℃、减压下对有机相进行2小时脱水,得到目标单体a3。所得到的单体a3的纯度为93.8质量%,二(甲基)丙烯酸酯单体量为6.2质量%。
[0059]
(实施例2-1)向装有搅拌机、温度计、冷凝器、滴液漏斗及氮气导入管的1l分离式烧瓶中加入350g的丙二醇单甲醚,对烧瓶内进行氮气置换,使其为氮气气氛。分别制备混合了80.0g的甲基丙烯酸正丁酯(mitsubishi gas chemical company,inc.制造)、80.0g的苯乙烯(ns styrene monomer co.,ltd.制造)、40.0g的单体a1及60g的丙二醇单甲醚的单体溶液,及混合了50g的丙二醇单甲醚和2.0g的2,2
’‑
偶氮双(2,4-二甲基戊腈)(产品名称:v-65(wako pure chemical industries,ltd.制造))的聚合引发剂溶液。
[0060]
将反应容器内升温至75℃,以3小时同时滴加单体溶液及聚合引发剂溶液。然后以75℃反应3小时,得到共聚物p1的丙二醇单甲醚溶液。所得到的溶液的固体成分浓度为30.1%。
[0061]
(实施例2-2)除了将单体溶液变更为50.0g的甲基丙烯酸正丁酯与60.0g的苯乙烯、90.0g的单体a1、60g的丙二醇单甲醚,并将2,2
’‑
偶氮双(2,4-二甲基戊腈)的量变更为4.0g以外,以与实施例1-1相同的方法得到共聚物p2。所得到的溶液的固体成分浓度为30.3%。
[0062]
(实施例2-3)除了将单体溶液变更为70.0g的甲基丙烯酸正丁酯、130.0g的单体a1、60g的丙二醇单甲醚,并将2,2
’‑
偶氮双(2,4-二甲基戊腈)的量变更为8.0g以外,以与实施例1-1相同的方法得到共聚物p3。所得到的溶液的固体成分浓度为29.9%。
[0063]
(实施例2-4)除了将单体溶液变更为80.0g的甲基丙烯酸正丁酯与30.0g的苯乙烯、90.0g的单体a2、60g的丙二醇单甲醚,并将2,2
’‑
偶氮双(2,4-二甲基戊腈)的量变更为4.0g以外,以与实施例1-1相同的方法得到共聚物p2。所得到的溶液的固体成分浓度为30.4%。
[0064]
(比较例2-1)除了将单体溶液变更为80.0g的甲基丙烯酸正丁酯与80.0g的苯乙烯、40.0g的单体a3、60g的丙二醇单甲醚以外,以与实施例1-1相同的方法得到共聚物p5。所得到的溶液的固体成分浓度为30.2%。
[0065]
(比较例2-2)除了将单体溶液变更为80.0g的甲基丙烯酸正丁酯与20.0g的苯乙烯、100.0g的单体a3、60g的丙二醇单甲醚以外,以与实施例1-1相同的方法合成共聚物p6,但在聚合中途发生了凝胶化。
[0066]
[共聚物的分析](聚合物的重均分子量(mw))通过凝胶浸透色谱(gpc)测定并根据下述条件求出。gpc装置:tosoh corporation制造,hlc-8220色谱柱:showa denko k.k.制造,shodex kf-805l溶剂:四氢呋喃标准物质:聚苯乙烯
[0067]
(聚合物溶液的固体成分浓度)在铝盘中秤量1g聚合物溶液,使用真空干燥机以120℃干燥30分钟。由干燥前后的
重量计算出固体成分浓度。
[0068]
[评价方法](粘度评价)对所得到的聚合物溶液进行粘度评价。具体而言,测定刚合成聚合物溶液后的粘度(c)、及在40℃的恒温槽中保管1个月后的粘度(d)。以“保管后的粘度/刚合成后的粘度(d/c)”表示两个粘度的比,通过以下基准进行评价。
◎
:粘度比(d/c)为1.0以上且小于1.02
○
:粘度比(d/c)为1.02以上且小于1.1
△
:粘度比(d/c)为1.1以上且小于1.3
×
:粘度比(d/c)为1.3以上或无法测定
[0069]
(固化膜硬度的评价)以使聚合物中的氨基量与环碳酸酯基量为等摩尔的方式,向10g聚合物溶液中加入3,3
’‑
二哌啶,制成均匀的溶液。将该溶液涂布在玻璃基板上,通过真空干燥去除溶剂。将其放入80℃的恒温槽中,加热3小时,得到膜厚为5μm的固化膜。对于所得到的固化膜,以jis k5600为基准,评价铅笔硬度。
[0070]
(固化膜的密合性的评价)以使聚合物中的氨基量与环碳酸酯基量为等摩尔的方式,向10g聚合物溶液中加入3,3
’‑
二哌啶,制成均匀的溶液。将该溶液涂布在玻璃基板上,通过真空干燥去除溶剂。将其放入80℃的恒温槽中,加热3小时,得到膜厚为5μm的固化膜。在所得到的固化膜上切出100块的切痕,并贴附cellotape(注册商标)(nichiban co.,ltd.制造),剥离cellotape(注册商标),通过块数及外观,评价密合性。将无剥离掉的块的评价为
“○”
,将无剥离掉的块但观察到碎屑的评价为
“△”
,将有剥离掉的块的评价为
“×”
。
[0071]
[表2]
[0072]
由表2可知,本发明的聚合物能够抑制经时性增稠,固化膜的硬度及密合性高。反之,比较例2-1的聚合物的经时性增稠大,铅笔硬度也低。并且,比较例2-2的聚合物发生凝胶化,未能形成固化膜。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1