γ-氨基丁酸(GABA)的氟取代的环己烯类似物
γ-氨基丁酸(gaba)的氟取代的环己烯类似物
1.对相关专利申请的交叉引用
2.本技术要求2019年10月25日提交的美国临时申请号62/926,120在35u.s.c.
§
119(e)下的优先权权益,其内容通过引用整体并入本文。
3.关于联邦资助研究或开发的声明
4.本发明是在美国国立卫生研究院(national institutes of health)授予的da030604下在政府支持下完成。政府享有本发明的某些权利。
5.背景
6.鸟氨酸氨基转移酶(oat)是一种5
′‑
磷酸吡哆醛(plp)依赖性的酶1,其存在于大多数人和动物组织的线粒体基质中2。oat催化在前半个反应中鸟氨酸向δ1-吡咯啉-5-羧酸(p5c)的转化和在后半个反应中α-酮戊二酸(α-kg)向谷氨酸的转化3。谷氨酸可以被谷氨酸脱氢酶4(glud)降解回α-kg或被吡咯啉-5-羧酸合酶5(p5cs)转化为p5c;通过oat或p5cs途径产生的p5c可以进一步转化为脯氨酸,这由吡咯啉-5-羧酸还原酶(pycr)介导
6-7
。由于脯氨酸通常沉积和储存在胶原中而不是游离氨基酸库中
7-8
,oat的活性与脯氨酸和胶原水平正相关9。胶原是细胞外基质(ecm)的主要组分
10
,并且胶原沉积或降解的变化可以导致ecm稳态的丧失。在过去几年中,ecm动力学和由ecm呈现出的化学信号的调节异常已被认为是发育和癌症进展的关键驱动因素
11
。此外,越来越清楚的是,脯氨酸代谢在癌症的代谢重编程中起着重要作用
12-13
。脯氨酸合成的酶被两种公认的癌基因c-myc和磷脂酰肌醇3-激酶(pi3k)显著上调
14
,从而导致作为中心中间体的p5c的合成上调
15
。oat将鸟氨酸转氨基以形成p5c是脯氨酸代谢和尿素循环之间的重要途径。发现oat在pk/c-myc转基因小鼠的肝肿瘤组织中被强烈活化
16
。在肝细胞癌(hcc)细胞中,oat基因的过表达是由wnt/β-连环蛋白途径的不适当活化诱导
16-17
。密切相关的脯氨酸代谢途径被hcc中的低氧微环境活化,其特征是促进肿瘤进展和索拉非尼抗性的羟基脯氨酸的积累
18
。oat的选择性抑制已被证明有效地抑制体内hcc肿瘤生长
19
。近年来,在非小细胞肺癌(nsclc)细胞中发现了oat的上调,其有助于增殖、侵袭和迁移的促进,细胞凋亡的抑制,以及细胞周期的改变
20
。此外,oat的敲低可以抑制nsclc细胞的体外增殖并抑制肺癌异种移植物模型中的体内肿瘤生长
20
。特异性oat敲低会阻断细胞分裂并造成人宫颈癌和骨肉瘤细胞的细胞死亡
21
。因此,oat的选择性抑制可以作为癌症治疗的潜在治疗策略。
技术实现要素:
7.公开了用于选择性抑制鸟氨酸氨基转移酶的化合物、组合物和相关使用方法。公开的化合物、组合物和方法可以用于治疗与鸟氨酸氨基转移酶活性有关的疾病和障碍。
8.公开的化合物可以被描述为被取代的环己烯化合物。具体地,公开的化合物可以被描述为氨基、氟取代的环己烯羧酸化合物。公开的化合物及其组合物可以用在调节鸟氨酸氨基转移酶(oat)活性的方法中,包括用于治疗与oat活性或表达有关的疾病或障碍诸如细胞增殖性疾病和障碍的方法。
9.公开的化合物可以涉及下式的化合物或其解离形式、非质子化形式、两性离子形
式或盐:
[0010][0011]
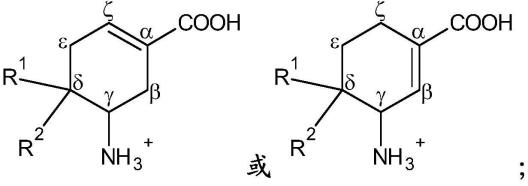
其中双键存在于α和ζ碳之间,或双键存在于α和β碳之间,且所述化合物具有下式:
[0012][0013]
其中r1和r2中的每一个独立地选自h或离去基团(例如,卤化物诸如f、cl、br和i),前提条件是,r1和r2中的至少一个不是h。
[0014]
在某些实施方案中,公开的化合物可以涉及下式的化合物或其解离形式、非质子化形式、两性离子形式或盐:
[0015][0016]
其中双键存在于α和ζ碳之间或α和β碳之间,
[0017]
其中r1和r2中的每一个独立地选自h或离去基团(例如,卤化物诸如f、cl、br和i),前提条件是,r1和r2中的至少一个不是h。
[0018]
在某些实施方案中,所述化合物是下式的化合物或其解离形式、非质子化形式、两性离子形式或盐,或这样的化合物的盐:
[0019][0020]
其中r1和r2中的每一个独立地选自h和f,前提条件是,r1和r2中的至少一个是f。
[0021]
在某些实施方案中,所述化合物是下式的化合物或其解离形式、非质子化形式、两性离子形式或盐,或这样的化合物的盐:
[0022][0023]
其中r1和r2中的每一个独立地选自h和f,前提条件是,r1和r2中的至少一个是f。
[0024]
如所指出的,公开的化合物可以被质子化,例如以形成铵部分,任选地在所述化合物作为盐存在的情况下。公开的化合物也可以是非质子化的和/或解离的,例如,其中羧酸部分被解离以形成羧酸根部分,任选地在所述化合物作为盐存在的情况下。公开的化合物可以是两性离子形式,其中所述化合物包含质子化的铵部分和解离的羧酸根部分,任选地在所述化合物作为盐存在的情况下。
[0025]
公开的化合物和组合物可以用在调节鸟氨酸氨基转移酶(oat)活性的方法中。这样的方法可以包含提供如在本文公开的化合物,诸如下式的化合物或其解离形式、两性离子形式或盐,或这样的化合物的盐,和使oat与所述化合物接触:
[0026][0027]
其中双键存在于α和ζ碳之间或α和β碳之间,
[0028]
其中r1和r2中的每一个独立地选自h和f,前提条件是,r1和r2中的至少一个不是h。在某些实施方案中,所述双键是在α和ζ碳之间。在某些实施方案中,所述双键是在α和β碳之间。在某些实施方案中,r1和r2是f。
[0029]
在某些实施方案中,公开的方法可以涉及降低癌症表达的oat的活性,所述癌症可以包括但不限于肝细胞癌(hcc)和非小细胞肺癌(nsclc)、或表达或过表达oat的其它癌症。这样的方法可以包含提供如在本文公开的化合物,诸如下式的化合物或其解离形式、非质子化形式、两性离子形式或盐,或这样的化合物的盐,和使所述癌症与所述化合物接触:
[0030][0031]
其中双键存在于α和ζ碳之间或α和β碳之间,
[0032]
其中r1和r2中的每一个独立地选自h和f,前提条件是,r1和r2中的至少一个不是h。在某些实施方案中,所述双键是在α和ζ碳之间。在某些实施方案中,所述双键是在α和β碳之间。在某些实施方案中,r1和r2是f。
[0033]
在某些实施方案中,公开的方法可以涉及在有此需要的受试者中治疗细胞增殖性疾病或障碍。合适的细胞增殖性疾病和障碍可以包括表达或过表达oat的癌症,诸如,但不限于肝细胞癌(hcc)和非小细胞肺癌(nsclc)。这样的方法可以包含给这样的有此需要的受
试者施用下式的化合物或其解离形式、非质子化形式、两性离子形式或盐,或这样的化合物的盐:
[0034][0035]
其中双键存在于α和ζ碳之间或α和β碳之间,
[0036]
其中r1和r2中的每一个独立地选自h和f,前提条件是,r1和r2中的至少一个不是h。在某些实施方案中,所述双键是在α和ζ碳之间。在某些实施方案中,所述双键是在α和β碳之间。在某些实施方案中,r1和r2是f。
[0037]
本文公开的化合物没有立体化学或构型限制并且包括所有立体化学或构型异构体,除非指出立体化学或构型限制。如下面说明和讨论的,这样的化合物和/或其中间体可以作为单一对映异构体、可以从中拆分异构体的外消旋混合物、或可以从中分离相应对映异构体的非对映异构体获得。因此,任何立构中心相对于任何其它立构中心可以是(s)或(r)。作为另一个单独的考虑,各种化合物可以作为酸或碱盐存在,部分地或完全地质子化,例如在氨基处,以形成铵部分,和/或部分地或完全地解离,例如在羧基处,以形成羧酸根取代基或部分。在某些这样的实施方案中,关于铵取代基或部分,抗衡离子可以是质子酸的共轭碱。在某些这样的或其它的实施方案中,关于羧酸根取代基或部分,抗衡离子可以是碱金属、碱土金属或铵阳离子。此外,本领域技术人员将理解,本文公开的任何一种或多种化合物可以作为药物组合物的一部分提供,所述药物组合物包含用于与治疗方法或药物结合使用的药学上可接受的载体组分。
[0038]
在某些实施方案中,公开的方法涉及与oat活性和/或表达或过表达有关的疾病或障碍,包括细胞增殖性疾病和障碍诸如与oat活性和/或表达或过表达有关的癌症。合适的疾病和障碍可以包括、但不限于细胞增殖性疾病和障碍,其可以包括、但不限于在需要这样的治疗的人受试者中的肝细胞癌(hcc)和非小细胞肺癌(nsclc)。在某些实施方案中,这样的化合物可以提供为药物组合物的一部分。
[0039]
在某些实施方案中,公开的方法涉及降低或调节由癌症(例如,肝细胞癌(hcc)和非小细胞肺癌(nsclc))表达的鸟氨酸氨基转移酶的活性。这样的方法可以包含提供上面讨论或本文别处描述的种类的化合物,和使这样的化合物与细胞培养基接触,所述细胞培养基包含表达鸟氨酸氨基转移酶的癌症,其中这样的化合物的量有效地降低鸟氨酸氨基转移酶活性。在某些实施方案中,这样的化合物可以提供为药物组合物的一部分。无论如何,这样的接触可以是在体外或在体内。
[0040]
更一般而言,公开的方法可以涉及抑制或钝化(inactivate)鸟氨酸氨基转移酶。这样的方法可以包含提供上文讨论或下文描述的种类的化合物,无论是否为药物组合物的一部分,和施用有效量的这样的化合物以与鸟氨酸氨基转移酶接触。如本领域所理解的,这样的接触可以用于实验和/或研究目的,或者可以设计为模拟一种或多种体内或生理条件。这样的化合物可以包括、但不限于由以下实施例、参考的附图、并入的参考文献和/或随附的合成方案说明的那些。在某些这样的实施方案中,这样的化合物和/或它们的组合可以以
至少部分地足以抑制oat、细胞增殖和/或肿瘤生长的量存在。
[0041]
附图简要描述
[0042]
图1.鸟氨酸的代谢途径。
[0043]
图2.选择性hoat钝化剂7和氟取代的环己烯类似物8-11的结构。
[0044]
图3.被8(左)和9(右)钝化的hoat的共晶结构。
[0045]
图4.9对鸟氨酸氨基转移酶(oat)的抑制活性。
[0046]
图5.9对γ-氨基丁酸氨基转移酶(gaba-at)的抑制活性。
[0047]
图6.9对丙氨酸氨基转移酶和天冬氨酸氨基转移酶的抑制活性。
[0048]
图7.通过不同浓度的8部分地或完全地抑制的hoat的时间依赖性透析。
[0049]
图8.通过不同浓度的9部分地或完全地抑制的hoat的时间依赖性透析。
[0050]
图9.使用基于机制的酶钝化剂滴定酶。将酶活性的损失测量为钝化与酶浓度之比的函数。对曲线的线性部分使用线性回归以获得x-截距,其为转换数(分配比=转换数-1)。确定8的转换数。
[0051]
图10.使用基于机制的酶钝化剂滴定酶。将酶活性的损失测量为钝化与酶浓度之比的函数。对曲线的线性部分使用线性回归以获得x-截距,其为转换数(分配比=转换数-1)。确定9的转换数。
[0052]
详细描述
[0053]
可以使用如下定义和术语进一步描述公开的主题。本文使用的定义和术语仅用于描述特定实施方案的目的,并不旨在进行限制。
[0054]
如在本说明书和权利要求书中所使用的,单数形式“一个/种(a)”、“一个/种(an)”和“所述(the)”包括复数形式,除非上下文另外清楚地指明。例如,术语“一个取代基”应当解释为是指“一个或多个取代基”,除非上下文另外清楚地指明。
[0055]
本文中使用的“约”、“大约”、“实质上”和“显著地”将被本领域普通技术人员理解,并且将在一定程度上根据使用它们的上下文而变化。如果该术语的使用对于本领域普通技术人员来说在使用它的上下文中是不清楚的,则“约”和“大约”将是指特定术语的至多
±
10%,且“实质上”和“显著地”将是指特定术语的超过
±
10%。
[0056]
本文中使用的术语“包括”具有与术语“包含”相同的含义。术语“包含”应当解释为“开放的”过渡性术语,其允许在权利要求中列举的那些组分之外进一步包含另外组分。术语“组成”和“由
……
组成”应当解释为“封闭的”过渡性术语,其不允许包含除权利要求中列举的组分之外的另外组分。术语“基本上由
……
组成”应当解释为部分地封闭,并且仅允许包括不会从根本上改变要求保护的主题的性质的另外组分。
[0057]
短语“诸如”应当解释为“例如,包括”。此外,任何和所有示例性语言(包括、但不限于“诸如”)的使用仅旨在更好地阐明要求保护的主题,而不是对要求保护的主题的范围构成限制。
[0058]
此外,在使用类似于“a、b和c等中的至少一个”的惯例的那些情况下,一般而言,这样的语法结构意在使本领域普通技术人员理解该惯例(例如,“具有a、b和c中的至少一个的系统”将包括、但不限于具有单独a、单独b、单独c、a和b一起、a和c一起、b和c一起、和/或a、b和c一起的系统)。本领域技术人员将进一步理解,无论是在说明书中还是在附图中,实际上呈现两个或更多个可替代术语的任何分离词和/或短语都应该被理解为涵盖包括术语之
一、术语中的任一个、或两个术语的可能性。例如,短语“a或b”将被理解为包括“a”或“b”或“a和b”的可能性。
[0059]
所有语言诸如“至多”、“至少”、“大于”、“小于”等都包括所列举的数字,并表示随后可以被分解为范围和子范围的范围。范围包括每个单独的成员。因此,例如,具有1-3个成员的集合表示具有1、2或3个成员的集合。类似地,具有6个成员的集合表示具有1、2、3、4或6个成员的集合,诸如此类。
[0060]
情态动词“可以(may)”表示在几个描述的实施方案或其中所含的特征之间的一种或多种选项或选择的优选使用或选择。在没有关于特定实施方案或其中所含的特征公开选项或选择的情况下,情态动词“可以(may)”表示关于如何制作或使用所描述的实施方案或其中所含的特征及其方面的肯定行为,或者使用关于所描述的实施方案或其中所含的特征的特定技能的确定性决策。在该后一种情况下,情态动词“可以(may)”具有与助动词“可以(can)”相同的含义和内涵。
[0061]
本文中使用的“有此需要的受试者”可以包括人和/或非人动物。“有此需要的受试者”可以包括具有与鸟氨酸氨基转移酶(oat)活性有关的疾病或障碍的受试者。“有此需要的受试者”可以包括具有细胞增殖性疾病或障碍的受试者,所述细胞增殖性疾病或障碍可以包括、但不限于肝细胞癌(hcc)。
[0062]
化学实体
[0063]
本文中公开了新的化学实体和化学实体的用途。可以使用本领域已知的术语来描述所述化学实体并且在下文进一步讨论。
[0064]
本文中使用的破折号
“‑”
、星号“*”或加号“+”可以用于指示任何自由基基团或取代基基团的连接点。
[0065]
本文所考虑的术语“烷基”包括所有其异构形式中的直链或支链烷基自由基,诸如1-12、1-10或1-6个碳原子的直链或支链基团,在本文中分别被称作c
1-c
12
烷基、c
1-c
10-烷基和c
1-c
6-烷基。
[0066]
术语“亚烷基”表示直链或支链烷基基团的二自由基(即,直链或支链c
1-c6烷基基团的二自由基)。示例性的亚烷基基团包括、但不限于-ch
2-、-ch2ch
2-、-ch2ch2ch
2-、-ch(ch3)ch
2-、-ch2ch(ch3)ch
2-、-ch(ch2ch3)ch
2-等。
[0067]
术语“卤代”表示卤素取代(例如,-f、-cl、-br或-i)。术语“卤代烷基”表示被至少一种卤素取代的烷基基团。例如,-ch2f、-chf2、-cf3、-ch2cf3、-cf2cf3等。
[0068]
本文中使用的术语“杂烷基”表示其中至少一个碳原子已被杂原子(例如,o、n或s原子)替代的“烷基”基团。一种类型的杂烷基基团是“烷氧基”基团。
[0069]
本文中使用的术语“烯基”表示具有至少一个碳-碳双键的不饱和直链或支链烃,诸如2-12、2-10或2-6个碳原子的直链或支链基团,在本文中分别被称作c
2-c
12-烯基、c
2-c
10-烯基和c
2-c
6-烯基。
[0070]
本文中使用的术语“炔基”表示具有至少一个碳-碳三键的不饱和直链或支链烃,诸如2-12、2-10或2-6个碳原子的直链或支链基团,在本文中分别被称作c
2-c
12-炔基、c
2-c
10-炔基和c
2-c
6-炔基。
[0071]
术语“环烷基”表示本文中提及的3-12、3-8、4-8或4-6个碳的单价饱和环状、二环或桥连环状(例如,金刚烷基)烃基,例如,从环烷烃衍生出的“c4-8-环烷基”。除非另有说
明,否则环烷基基团任选地在一个或多个环位置处被例如烷酰基、烷氧基、烷基、卤代烷基、烯基、炔基、酰氨基或羧基酰氨基、脒基、氨基、芳基、芳基烷基、叠氮基、氨基甲酸酯、碳酸酯、羧基、氰基、环烷基、酯、醚、甲酰基、卤素、卤代烷基、杂芳基、杂环基、羟基、亚氨基、酮、硝基、磷酸酯、磷酸根合(phosphonato)、次膦酸根合(phosphinato)、硫酸酯、硫化物、磺酰氨基、磺酰基或硫代羰基取代。在某些实施方案中,所述环烷基基团未被取代,即,它是未被取代的。
[0072]
术语“环杂烷基”表示3-12、3-8、4-8或4-6个碳的单价饱和环状、二环或桥连环状烃基,其中环烷烃的至少一个碳被杂原子(例如,n、o和/或s)替代。
[0073]
术语“亚环烷基”表示在一个或多个环键处不饱和的环烷基基团。
[0074]
术语“部分不饱和碳环基”表示在环原子之间含有至少一个双键的单价环状烃,其中碳环基的至少一个环不是芳族的。部分不饱和碳环基可以根据环碳原子的数目来表征。例如,部分不饱和碳环基可以含有5-14、5-12、5-8或5-6个环碳原子,并因此分别被称作5-14、5-12、5-8或5-6元部分不饱和碳环基。部分不饱和碳环基可以呈单环碳环、二环碳环、三环碳环、桥连碳环、螺环碳环或其它碳环环系统的形式。示例性的部分不饱和碳环基基团包括部分不饱和的环烯基基团和二环碳环基基团。除非另有说明,否则部分不饱和碳环基基团任选地在一个或多个环位置处被例如烷酰基、烷氧基、烷基、卤代烷基、烯基、炔基、酰氨基或羧基酰氨基、脒基、氨基、芳基、芳基烷基、叠氮基、氨基甲酸酯、碳酸酯、羧基、氰基、环烷基、酯、醚、甲酰基、卤素、卤代烷基、杂芳基、杂环基、羟基、亚氨基、酮、硝基、磷酸酯、磷酸根合(phosphonato)、次膦酸根合(phosphinato)、硫酸酯、硫化物、磺酰氨基、磺酰基或硫代羰基取代。在某些实施方案中,所述部分不饱和碳环基未被取代,即,它是未被取代的。
[0075]
术语“芳基”是本领域公认的,并且表示碳环的和/或杂环的芳族基团。代表性的芳基基团包括苯基、萘基、蒽基、吡啶基、喹啉基、呋喃基、亚硫酰基等。术语“芳基”包括具有两个或更多个碳环的多环环系统,其中两个或更多个碳是两个邻接环所共有的(环是“稠合环”),其中环中的至少一个是芳族的,且例如,其它环可以是环烷基、环烯基、环炔基和/或芳基。除非另有说明,否则芳族环可以在一个或多个环位置处被例如卤素、叠氮化物、烷基、芳烷基、烯基、炔基、环烷基、羟基、烷氧基、氨基、硝基、巯基、亚氨基、酰氨基或羧基酰氨基、羧酸、-c(o)烷基、-co2烷基、羰基、羧基、烷基硫基、磺酰基、磺酰氨基、磺酰胺、酮、醛、酯、杂环基、芳基或杂芳基部分、-cf3、-cn等取代。在某些实施方案中,所述芳族环在一个或多个环位置处被卤素、烷基、羟基或烷氧基取代。在某些其它实施方案中,所述芳族环未被取代,即,它是未被取代的。在某些实施方案中,所述芳基基团是6-10元环结构。
[0076]
术语“杂环基”和“杂环基团”是本领域公知的,且表示饱和的、部分不饱和的或芳族的3-10元环结构,可替换地表示3-7元环,其环结构包括1-4个杂原子,诸如氮、氧和硫。杂环基基团中的环原子的数目可以使用5cx-cx命名法指定,其中x是指定环原子数目的整数。例如,c
3-c7杂环基基团表示含有1-4个杂原子(诸如氮、氧和硫)的饱和或部分不饱和3-7元环结构。命名“c
3-c
7”指示,杂环含有共3-7个环原子,包括占据环原子位置的任何杂原子。
[0077]
术语“胺”和“氨基”是本领域公知的,且表示未被取代的和被取代的胺(例如,单取代的胺或二取代的胺),其中取代基可以包括,例如,烷基、环烷基、杂环基、烯基和芳基。
[0078]
术语“烷氧基(alkoxy)”或“烷氧基(alkoxyl)”是本领域公知的,且表示具有与其相连的氧自由基的如上定义的烷基。代表性的烷氧基基团包括甲氧基、乙氧基、叔丁氧基
等。
[0079]“醚”是由氧共价连接的两个烃。因此,使烷基成为醚的烷基取代基是或类似于烷氧基,诸如可以以-o-烷基、-o-烯基、-o-炔基等之一为代表。
[0080]
本文中使用的术语“羰基”表示自由基-c(o)-。
[0081]
术语“氧代”表示二价氧原子-o-。
[0082]
本文中使用的术语“羧基(carboxy)”或“羧基(carboxyl)”表示自由基-cooh或它的相应盐,例如-coona等。羧基烷基酯表示具有部分-c(o)o-r的化合物,其中r是烷基。
[0083]
本文中使用的术语“甲酰氨基”表示自由基-c(o)nrr',其中r和r'可以相同或不同。例如,r和r'可以独立地是氢、烷基、芳基、芳基烷基、环烷基、甲酰基、卤代烷基、杂芳基或杂环基。
[0084]
本文中使用的术语“酰胺”或“酰氨基”或“酰胺基”表示形式-r1c(o)n(r2)-、-r1c(o)n(r2)r
3-、-c(o)nr2r3或-c(o)nh2的自由基,其中r1、r2和r3例如各自独立地是氢、烷基、烷氧基、烯基、炔基、酰胺、氨基、芳基、芳基烷基、氨基甲酸酯、环烷基、酯、醚、甲酰基、卤素、卤代烷基、杂芳基、杂环基、氢、羟基、酮或硝基。
[0085]
本公开内容的化合物可以是异构的。在某些实施方案中,公开的化合物可以是异构纯的,其中所述化合物占化合物的异构混合物内的所有化合物的大于约99%。本文也涵盖组合物,其包含异构纯的化合物和/或异构富集的组合物、基本上由其组成或由其组成,所述组合物可以包含至少约50%、60%、70%、80%、90%、95%、96%、97%、98%、99%或100%的给定化合物的单一异构体,基本上由其组成或由其组成。
[0086]
本公开内容的化合物可以含有一个或多个手性中心和/或双键,且因此,作为立体异构体诸如几何异构体、对映异构体或非对映异构体存在。当在本文中使用时,术语“立体异构体”由所有的几何异构体、对映异构体或非对映异构体组成。这些化合物可以用符号“r”或“s”、或“+”或
“‑”
表示,取决于在手性或立体异构源碳原子周围的取代基的构型和/或观察到的旋光度。公开的化合物包括各种立体异构体及其混合物。立体异构体包括对映异构体和非对映异构体。对映异构体或非对映异构体的混合物在命名法中可以用(
±
)表示,但熟练的技术人员会认识到,结构可以隐含地表示手性中心。应当理解,化学结构的图解描绘(例如,通用化学结构)包括所指化合物的全部立体异构形式,除非另外指出。本文也涵盖组合物,其包含对映异构纯的化合物和/或对映异构富集的组合物、基本上由其组成或由其组成,所述组合物可以包含至少约50%、60%、70%、80%、90%、95%、96%、97%、98%、99%或100%的给定化合物的单一对映异构体(例如,至少约95%的给定化合物的r对映异构体),基本上由其组成或由其组成。
[0087]
在理解oat的催化机制以及gaba-at和oat的钝化机制的情况下,可以考虑公开的化合物和使用方法的各种非限制性实施方案。在某些实施方案中,公开的主题涉及一种或多种如上所述的oat抑制剂,其与一种或多种生理上可耐受或可接受的稀释剂、载体、辅助剂或媒介物(它们在本文中统称为载体)一起配制成组合物。适合用于这样的接触或施用的组合物可以包含生理上可接受的水性或非水性性溶液、分散体、混悬剂或乳剂,无论是否是无菌的。所得组合物可以与本文所述的各种方法结合用于施用或接触鸟氨酸氨基转移酶。无论是否与药物组合物结合,“接触”是指,将鸟氨酸氨基转移酶与一种或多种抑制剂化合物放在一起,目的是使这样的抑制剂化合物与酶结合和/或络合。有效抑制鸟氨酸氨基转移
酶的化合物的量可以凭经验确定,并且做出这样的确定是在本领域的技术范围内。抑制或以其它方式影响鸟氨酸氨基转移酶活性包括降低、减轻和/或调节、以及消除oat活性、谷氨酸产生、谷氨酰胺合成、细胞增殖和/或肿瘤生长。
[0088]
本领域技术人员应理解,剂量将随特定抑制剂化合物的活性、疾病状态、施用途径、治疗持续时间以及医学和药学领域众所周知的类似因素而变化。一般而言,合适的剂量将是这样的量:其为有效产生治疗或预防效果的最低剂量。如果需要的话,可以将有效剂量的这样的化合物、其药学上可接受的盐或相关组合物以两个或更多个亚剂量施用,在适当的时间段内分别施用。
[0089]
制备药物制剂或组合物的方法包括使抑制剂化合物与载体和任选的一种或多种额外的辅助剂或成分结合的步骤。例如,可以采用标准药物制剂技术,诸如在remington's pharmaceutical sciences,mack publishing company,easton,pa中描述的那些。
[0090]
无论组合物或制剂如何,本领域技术人员会认识到用于药物施用的各种途径,以及在使这样的药物适合于施用时要考虑的相应因素和参数。因此,关于一个或多个非限制性实施方案,公开的化合物可以用作用于制备药物的抑制剂化合物,所述药物用于治疗或预防与oat活性、表达或过表达相关的疾病或障碍的治疗用途。合适的疾病或障碍可以包括细胞增殖性疾病或障碍,其可以包括、但不限于肝细胞癌(hcc)和非小细胞肺癌(nsclc)。
[0091]
通常,关于各种实施方案,公开的主题可以涉及用于治疗病理性增殖性障碍的方法。本文中使用的术语“障碍”表示其中存在正常功能紊乱的病症。“疾病”是对受影响的人或与该人接触的人造成不适、功能障碍或痛苦的身体或精神的任何异常病症。有时该术语被广泛使用以包括损伤、残疾、综合征、症状、异常行为以及结构和功能的非典型变化,而在其它上下文中,这些可能被认为是可辨别的类别。应当指出,术语“疾病”、“障碍”、“病症”和“病”在本文中同样地使用。
[0092]
根据某些实施方案,所公开的方法可以特别适用于治疗恶性增殖性障碍,包括表达鸟氨酸氨基转移酶(oat)的恶性增殖性障碍。本文中使用的“癌症”、“肿瘤”和“恶性肿瘤”都等同地涉及组织或器官的增生。如果组织是淋巴或免疫系统的一部分,恶性细胞可以包括循环细胞的非实体瘤。其它组织或器官的恶性肿瘤可能产生实体瘤。因此,本文公开的化合物、组合物和方法可以用于治疗非实体瘤和实体瘤。
[0093]
本文所考虑的恶性肿瘤可以选自表达oat的黑素瘤、癌、白血病、淋巴瘤和肉瘤。可以通过本文公开的方法治疗的恶性肿瘤(包括表达oat的恶性肿瘤)可以包含、但不限于血液学恶性肿瘤(包括白血病、淋巴瘤和骨髓增生性障碍)、再生不良性和再生障碍性贫血(病毒诱导的和特发性的)、骨髓增生异常综合征、所有类型的副肿瘤综合征(免疫介导的和特发性的)和实体瘤(包括膀胱、直肠、胃、子宫颈、卵巢、肾、肺、肝、乳房、结肠、前列腺、胃肠道、胰腺和卡波西)。更具体地,根据某些实施方案,联合使用的化合物和组合物可以用于治疗或抑制非实体癌症的方法中,所述非实体癌症是例如造血系统恶性肿瘤诸如所有类型的白血病,例如急性淋巴细胞白血病(all)、急性髓细胞性白血病(aml)、慢性淋巴细胞白血病(cll)、慢性髓细胞性白血病(cml)、骨髓增生异常综合征(mds)、肥大细胞白血病、毛细胞白血病、霍奇金病、非霍奇金淋巴瘤、伯基特氏淋巴瘤和多发性骨髓瘤,以及用于治疗或抑制实体瘤诸如唇和口腔、咽、喉、鼻旁窦、大唾液腺、甲状腺、食管、胃、小肠、结肠、结直肠、肛管、肝、胆囊、肝外(extraliepatic)胆管、法特壶腹、外分泌胰腺、肺、胸膜间皮瘤、骨中的肿
瘤,软组织肉瘤,皮肤癌和恶性黑素瘤,乳房、外阴、阴道、子宫颈、子宫体、卵巢、输卵管、滋养细胞肿瘤,阴茎、前列腺、睾丸、肾、肾盂、输尿管、膀胱、尿道中的肿瘤,眼睑癌,结膜癌,结膜的恶性黑素瘤,恶性黑素瘤,视网膜母细胞瘤,泪腺癌,眼眶肉瘤,脑、脊髓、血管系统、血管肉瘤和卡波西氏肉瘤。
[0094]
本文中公开的化合物和组合物可以以本领域已知的治疗方法施用。因此,各种这样的化合物和组合物可以以任何合适的方式与这样的方法结合施用。例如,施用可以包含口服、静脉内、动脉内、肌肉内、皮下、腹膜内、胃肠外、透皮、阴道内、鼻内、粘膜、舌下、局部、直肠或皮下施用或其任意组合。
[0095]
根据某些实施方案,治疗的受试者可以是哺乳动物受试者。尽管本文公开的方法特别旨在用于治疗人类的增殖性障碍,但也包括其它哺乳动物。作为非限制性例子,哺乳动物受试者包括猴、马属动物、牛、犬科动物、猫科动物、小鼠、大鼠和猪。
[0096]
如本文中和权利要求书中使用的术语“治疗”是指改善具有病理性障碍的受试者中的疾病活动性的一种或多种临床指标。“治疗”表示治疗性治疗。需要治疗的对象是遭受任何病理性障碍的哺乳动物受试者。“患者”或“有需要的受试者”是指,为了预防、克服、调节或减慢这样的伤害(infliction),期望施用本文所述种类的化合物或任何药物组合物的任何哺乳动物。提供“预防性治疗”或“预防治疗”是以保护方式起作用,以防御或预防某事,尤其是病症或疾病。
[0097]
更一般而言,所公开的方法可以涉及影响、调节、减少、抑制和/或预防与oat活性有关的恶性病理性增殖性障碍的起始、进展和/或转移(例如,从肝脏其它地方或从任何其它器官或组织到肝脏)(参见,例如,lucero om,dawson dw,moon rt,等人.a re-evaluation of the“oncogenic”nature of wnt/beta-catenin signaling in melanoma and other cancers.curr oncol rep 2010,12,314-318;liu wei;le anne;hancock chad;lane andrew n;dang chi v;fan teresa w-m;phang james m.reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-myc.proc.natl.acad.sci.usa 2012,109(23),8983-8988;和tong,xuemei;zhao,fangping;thompson,craig b.the molecular determinants of de novo nucleotide biosynthesis in cancer cells.curr.opin.genet.devel.2009,19(1),32-37.)。
[0098]
示例性实施方案
[0099]
以下实施方案是示例性的,并且不应当解释为限制要求保护的主题的范围。
[0100]
实施方案1.下式的化合物或其解离形式、两性离子形式或盐:
[0101][0102]
其中双键存在于α和ζ碳之间或α和β碳之间,且
[0103]
其中r1和r2中的每一个独立地选自h或离去基团诸如f、cl、br和i,前提条件是,r1和r2中的至少一个不是h。
[0104]
实施方案2.实施方案1的化合物,所述化合物呈包含铵部分和羧酸根部分的两性离子形式。
[0105]
实施方案3.实施方案1或2的化合物,其中所述双键是在α和ζ碳之间。
[0106]
实施方案4.实施方案1或2的化合物,其中所述双键是在α和β碳之间。
[0107]
实施方案5.前述实施方案中的任一项的化合物,其中r1和r2中的至少一个是f。
[0108]
实施方案6.前述实施方案中的任一项的化合物,其中所述化合物是包含取代基的盐,所述取代基选自铵取代基、羧酸根取代基和它们的组合。
[0109]
实施方案7.实施方案6的化合物,其中所述铵盐具有作为质子酸的共轭碱的抗衡离子。
[0110]
实施方案8.在药物组合物中的前述实施方案中的任一项的化合物,所述药物组合物包含药学上可接受的载体组分。
[0111]
实施方案9.具有下式的前述实施方案中的任一项的化合物或其盐:
[0112]
和任选地
[0113]
其中r1和r2中的每一个独立地选自h和f,前提条件是,r1和r2中的至少一个是f,且所述化合物任选地具有下式
[0114][0115]
实施方案10.实施方案9的化合物,其中r1和r2中的每一个是f。
[0116]
实施方案11.实施方案9的化合物,其中所述化合物是包含取代基的盐,所述取代基选自铵取代基、羧酸根取代基和它们的组合。
[0117]
实施方案12.实施方案11的化合物,其中所述铵盐具有作为质子酸的共轭碱的抗衡离子。
[0118]
实施方案13.在药物组合物中的实施方案9的化合物,所述药物组合物包含药学上可接受的载体组分。
[0119]
实施方案14.具有下式的前述实施方案中的任一项的化合物或其盐:
[0120]
和任选地
[0121]
其中r1和r2中的每一个独立地选自h和f,前提条件是,r1和r2中的至少一个是f,且所述化合物任选地具有下式
[0122][0123]
实施方案15.实施方案14的化合物,其中r1和r2中的每一个是f。
[0124]
实施方案16.实施方案14的化合物,其中所述化合物是包含取代基的盐,所述取代基选自铵取代基、羧酸根取代基和它们的组合。
[0125]
实施方案17.实施方案16的化合物,其中所述铵盐具有作为质子酸的共轭碱的抗衡离子。
[0126]
实施方案18.在药物组合物中的实施方案14的化合物,所述药物组合物包含药学上可接受的载体组分。
[0127]
实施方案19.一种药物组合物,其包含:(i)前述实施方案中的任一项的化合物;和(ii)药学上合适的载体、稀释剂或赋形剂。
[0128]
实施方案20.一种药物组合物,其包含:(i)实施方案9的化合物;和(ii)药学上合适的载体、稀释剂或赋形剂。
[0129]
实施方案21.一种药物组合物,其包含:(i)实施方案14的化合物;和(ii)药学上合适的载体、稀释剂或赋形剂。
[0130]
实施方案22.一种调节鸟氨酸氨基转移酶(oat)活性的方法,所述方法包含:(i)任选地提供权利要求1-18中的任一项的化合物;和(ii)使权利要求1-18中的任一项的化合物与包含oat的培养基接触,所述化合物的量足以调节oat活性。
[0131]
实施方案23.实施方案22的方法,其中所述双键是在α和ζ碳之间。
[0132]
实施方案24.实施方案22的方法,其中所述双键是在α和β碳之间。
[0133]
实施方案25.实施方案22-24中的任一项的方法,其中r1和r2中的至少一个是f。
[0134]
实施方案26.实施方案22-25中的任一项的方法,其中所述化合物是包含取代基的盐,所述取代基选自铵取代基、羧酸根取代基和它们的组合。
[0135]
实施方案27.实施方案26的方法,其中所述铵盐具有作为质子酸的共轭碱的抗衡离子。
[0136]
实施方案28.实施方案22-27中的任一项的方法,其中所述接触是在体内。
[0137]
实施方案29.一种降低人癌症表达的oat的活性的方法,所述方法包含:(i)任选地提供权利要求1-18中的任一项的化合物;和(ii)使权利要求1-18中的任一项的化合物与细胞培养基接触,所述细胞培养基包含表达oat的癌症,所述化合物的量有效地降低oat活性。
[0138]
实施方案30.实施方案29的方法,其中所述双键是在α和ζ碳之间。
[0139]
实施方案31.实施方案29的方法,其中所述双键是在α和β碳之间。
[0140]
实施方案32.实施方案29-31中的任一项的方法,其中r1和r2中的至少一个是f。
[0141]
实施方案33.实施方案29-32中的任一项的方法,其中所述化合物提供在药物组合物中。
[0142]
实施方案34.实施方案29-33中的任一项的方法,其中所述接触是在体内。
[0143]
实施方案35.实施方案29-33中的任一项的方法,其中所述接触是与有此需要的人
受试者接触。
[0144]
实施方案36.一种用于在有此需要的受试者中治疗癌症的方法,所述方法包含给所述受试者施用治疗有效量的实施方案1-18中的任一项的化合物。
[0145]
实施方案37.实施方案36的方法,其中所述双键是在α和ζ碳之间。
[0146]
实施方案38.实施方案36的方法,其中所述双键是在α和β碳之间。
[0147]
实施方案39.实施方案36-38中的任一项的方法,其中r1和r2中的至少一个是f。
[0148]
实施方案40.实施方案36-39中的任一项的方法,其中所述癌症特征在于鸟氨酸氨基转移酶(oat)的表达或过表达。
[0149]
实施方案41.实施方案36-40中的任一项的方法,其中所述癌症是肝细胞癌(hcc)。
[0150]
实施方案42.实施方案36-41中的任一项的方法,其中所述癌症是非小细胞肺癌(nsclc)。
实施例
[0151]
以下实施例是示例性的,且不应当解释为限制要求保护的主题的范围。以下非限制性实施例和数据说明了与公开的化合物、组合物和方法相关的各个方面和特征,包括治疗与oat活性、表达或过表达有关的疾病和障碍,和/或降低鸟氨酸氨基转移酶活性,所述疾病和障碍诸如细胞增殖性疾病和障碍,包括、但不限于肝细胞癌(hcc)和非小细胞肺癌(nsclc)。虽然通过几种化合物和组合物(可以以此使用)的应用来说明本发明的实用性,但是本领域技术人员将理解,用与本发明的范围相称的各种其它化合物可获得可比较的结果。
[0152]
实施例1
–
一个氟原子赋予γ-氨基丁酸的两种环己烯类似物对人鸟氨酸氨基转移酶的钝化机制的显著差异
[0153]
参考著作zhu,wei等人.“a remarkable difference that one fluorine atom confers on the mechanisms of inactivation of human ornithine aminotransferase by two cyclohexene analogues ofγ-aminobutyric acid.”j.am.chem.soc.第142卷,10:4892-4903,2020年3月1日,其内容通过引用整体并入。
[0154]
摘要
[0155]
人鸟氨酸氨基转移酶(hoat)(一种5-磷酸吡哆醛依赖性酶)在肝细胞癌(hcc)的进展中起关键作用。hoat的药理学选择性抑制已被证明是hcc的潜在治疗方案。受非选择性氨基转移酶钝化剂(1r,3s,4s)-3-氨基-4-氟环戊烷-1-甲酸(1)的发现的启发,我们在这项工作中合理设计、合成和评价了一系列新的氟取代的环己烯类似物,从而将8和9鉴定为新型选择性hoat时间依赖性抑制剂。完整蛋白质谱法(intact protein mass spectrometry)和蛋白晶体学证明8和9作为hoat的共价抑制剂,它们表现出由单个氟原子的差异导致的两种不同的钝化机制。令人感兴趣的是,根据基于质谱法的代谢物分析和氟离子释放实验,它们具有相似的转换机制。进行了分子动力学(md)模拟和静电势(esp)电荷计算,其阐明了单氟差异对相应中间体的显著影响,从而导致两种完全不同的钝化途径。9的新型加成芳构化(additionaromatization)钝化机制有助于其显著增强的效力,以及优秀的选择性(相对于其它氨基转移酶)。
[0156]
引言
[0157]
肝细胞癌(hcc)占原发性肝癌的90%,是全球第二大最常见的癌症死亡原因
1-4
。迄今为止,hcc没有有效治疗,因为它通常在晚期疾病阶段才被诊断,并且肿瘤通常难以用护理标准(standard-of-care)受体酪氨酸激酶抑制剂索拉非尼和放射疗法进行全身治疗
5-8
。人鸟氨酸氨基转移酶(hoat)是一种5
′‑
磷酸吡哆醛(plp)依赖性酶9,其在先天性代谢缺陷
10
和肝细胞癌(hcc)进展
11
中起作用。在机制上,两个偶联的半反应参与hoat的氨基转移循环(图1)。在前半个反应中,hoat催化plp和鸟氨酸向磷酸吡多胺(pmp)和谷氨酰基-5-半醛的转化,后者自发环化为δ1-吡咯啉-5-羧酸(p5c)
12
。生成的p5c可以通过吡咯啉-5-羧酸还原酶(pycr)进一步转化为脯氨酸
13,14
。在后半个反应中,pmp和α-酮戊二酸(α-kg)被转化为plp和l-谷氨酸(l-glu),后者也可以被吡咯啉-5-羧酸合酶(p5cs)转化为p5c。越来越多的证据表明,脯氨酸代谢在代谢重编程以维持癌症增殖中以及上调作为中心中间体的p5c的合成中起重要作用
15-17
。在hcc中的代谢重编程的特征在于脯氨酸/羟基脯氨酸代谢途径的活化,这支持hif1a依赖性的肿瘤进展和索拉非尼抗性
18
。此外,由hoat产生的谷氨酸可以通过谷氨酰胺合酶(gs)转化为谷氨酰胺(图1),这支持合成代谢和增殖性细胞程序
19
。发现hoat和谷氨酰胺生成酶(glutaminogenic enzyme)被强烈活化,并且因为异常的致癌wnt/β-连环蛋白信号传递而通常在hcc中过表达
20,21
。hoat的选择性抑制已被证明有效地抑制体内hcc肿瘤生长
11
。最近,还发现hoat的特异性敲低会在体外和在体内抑制非小细胞肺癌(nsclc)的生长
22
。总之,hoat通过脯氨酸和谷氨酰胺代谢途径在hcc的代谢重编程中发挥重要作用,并且hoat的选择性抑制是有前途的治疗hcc和其它相关癌症的治疗策略。在14种已知的氨基转移酶中,hoat与γ-氨基丁酸氨基转移酶(gaba-at)属于同一亚组,由于它们在一级结构中的相似性
23
。hoat和gaba-at具有非常相似的活性部位
24
,因此gaba-at的一些强效抑制剂也抑制hoat就不足为奇
11
。多年来,我们实验室一直专注于gaba-at的基于机制的钝化剂(mbi)的合理设计
25
。mbi是靶酶的非反应性替代底物,它们在催化部位转化为活性物质,并然后通过与酶的共价键合、紧密结合抑制或任何在功能上不可逆的抑制机制导致钝化
26
。例如,据报道,(1r,3s,4s)-3-氨基-4-氟环戊烷-1-甲酸(1)通过相同的烯胺机制钝化hoat
27
和gaba-at
28
,如在方案1中所示。所提出的钝化机制是由希夫碱2的形成引发,希夫碱2经受决定速率的去质子化和氟离子的原位消除以提供中间体4。活性烯胺5通过希夫碱交换反应形成,并且随后5向lys结合的plp复合物的亲核加成产生共价加合物6。
[0158]
方案1.1对hoat和gaba-at的钝化机制
[0159][0160]
因为mbi在它们的活性部位被活化之前是惰性的,因此可以大大减少不希望的脱靶效应。更重要的是,即使在较低剂量,这些钝化剂也可以表现出比传统抑制剂更高的效力和选择性
11,29,30
。在2015年,gaba类似物7(图2)被报道为hoat的选择性mbi,其在0.1和1.0mg/kg的剂量显著降低甲胎蛋白水平(hcc的生物标志物)并抑制体内hcc肿瘤生长
11
。最近,还揭示了7的钝化机制
31
。
[0161]
我们一直有兴趣于发现具有新颖钝化机制的有效的和选择性的hoat钝化剂。在此,我们在基于环戊烷的类似物1的基础上合理设计和合成了氟取代的基于环己烯的gaba类似物8-11(图2)。在它们中,化合物(9)作为hoat的钝化剂的效率是化合物7的23倍,并且具有优秀的选择性(相对于其它氨基转移酶(例如,gaba-at))。我们还通过质谱法、晶体学和各种其它生化方法阐明了8和9的钝化和转换机制,并可以得出结论,单个氟原子的差异会显著改变钝化机制。
[0162]
结果和讨论
[0163]
新型oat钝化剂的设计。在hoat和gaba-at中观察到类似的结合口袋,它们在配体的锚点之间产生几乎相同的距离。令人感兴趣的是,hoat中的tyr55部分地占据与gaba-at中的phe351相似的立体空间。疏水性更强的phe351残基侵入gaba-at的活性部位,使得只有较小的gaba分子可以适合;hoat催化部位是更柔性的和更大的,以容纳比gaba长一个碳的鸟氨酸
32
。此外,独特的tyr55残基提供了与hoat的活性部位中更亲水的底物形成额外氢键的潜力
31
。在以前,基于环戊烷的gaba类似物1被鉴定为针对hoat和gaba-at的双重抑制剂。由于1的相对小尺寸,它可以放入hoat和gaba-at的催化口袋中,这与其差的选择性一致
27
。因此,我们假设相对较大尺寸的环己烯衍生物8-11可能提高它们的hoat选择性(相对于gaba-at)(图2)。此外,与双键异构体10/11相比,双键在α/β-位置的引入(8/9)可能会增强抑制活性
33
,这可能是通过增加与氨基相邻的质子的酸度来实现。
[0164]
mbi通常最初充当底物,随后在催化部位形成活性中间体,其然后通过不同的机制途径使酶钝化。因此,钝化机制的提出在潜在mbi的合理设计的早期阶段起着重要作用。在方案2中提出了8/9的三种可能的钝化机制。所有的机制都由希夫碱形成(12a/12b)引发,随后hf消除以产生反应性物质14a/14b,这基于1的已知机制(方案1)。机制途径a类似于1的钝化机制,其中活性烯胺15a/15b可以在14a/14b与lys292的转亚胺作用(transimination)后释放。随后的烯胺添加可以导致钝化和加合物16a/16b的形成。可以进一步释放加合物16b的第二个氟离子以提供芳族加合物17。机制途径b是14a/14b的直接芳构化以产生紧密结合的加合物20a/20b,这类似于另一种氟代的环己烯类似物的钝化机制
34
。机制途径c涉及lys292向共轭烯烃14a/14b的亲核加成,产生加合物21a/21b。
[0165]
方案2.8和9的可能钝化机制
[0166][0167]
在21b的情况下,可能在消除第二个氟离子后形成更稳定的共价芳族加合物(26)。
[0168]
分子对接。为了更好地理解环大小之间的位阻的差异,采用1和8的分子对接研究
35
来模拟它们在gaba-at和hoat的活性部位处的结合位姿(与天然底物相比)。基于分子对接研究,gaba结合在gaba-at的活性部位并与残基arg192、tyr69和glu270建立氢键(数据未显示)。gaba的这种推定结合位姿将其γ-氨基基团定位在靠近lys329-plp复合物的位置并促进随后的希夫碱交换。1的结合模型展示出与这些残基相似的氢键(数据未显示)。但是,分子对接显示8与his206形成更稳定的氢键以避免与残基phe351的潜在冲突(数据未显示),
这可能阻碍其与lys329-plp复合物的初始反应。相比之下,鸟氨酸与hoat的结合模型显示δ-氨基基团深入活性部位并与thr322形成明显的氢键(数据未显示),这使其接近lys292-plp复合物。8的氨基基团在对接模型中与thr322形成类似的氢键(数据未显示),而1的对接位姿表明它反而与glu325形成氢键(数据未显示)。这些对接结果指示,与1相比,较大的环己烯类似物作为底物在hoat活性部位受到青睐,但在gabaat活性部位不受欢迎,这与我们上述的设计策略相匹配。
[0169]
发现mbi的γ-位置的手性对于钝化过程和保持其抑制活性非常重要
25
。令人感兴趣的是,8的对映异构体在hoat的活性部位显示出相似的结合位姿(数据未显示)。lys辅助的在γ-位置的去质子化被证明是早期类似物中的决定速率的步骤
36
。因此,还进行了分子对接研究以预测活性中间体12a(数据未显示)和它的对映异构体(数据未显示)在hoat的催化口袋处的结合位姿。这些中间体的羧酸根部分都与tyr55和arg180建立了氢键,而12a-对映异构体的不同手性迫使其γ-质子面向另一侧,其指向远离催化性lys292(vs )。该对接模拟也与早期钝化剂的观察一致,并支持手性纯类似物的合成。
[0170]
氟取代的环己烯类似物8-11的合成。8-11的合成途径如方案3所示。二-pmb中间体(29)从手性纯的起始原料27通过三个连续步骤提供:分子内环化、立体选择性环氧化物开环和用过量茴香醛进行还原胺化。将得到的中间体(29)用xtalfluor-m和(hf)3et3n处理,以中等收率排它地产生反式异构体30,其中可能涉及氮杂环丙烷鎓机制途径
37
。从二-pmb保护的中间体30至boc-保护的中间体31的直接转化是在有boc2o存在下通过pd(oh)2催化的氢化实现。将中间体31用phsecl和khmds处理,随后用m-cpba进行氧化消除以产生烯烃异构体32a和32b,将它们通过色谱法分离,并然后分别去保护为8和10。基于之前的报道,可以从手性纯的27制备醇33
38
;然后用pcc氧化33,以产生酮34。在筛选各种氟化试剂和反应温度以后,关键中间体35以中等收率得到,避免了单氟烯烃杂质的产生
39
。使用与用于制备8和10相同的方法,将中间体35转化为二氟烯烃异构体36a和36b,将它们分别进一步去保护为9和11。
[0171]
方案3.氟取代的环己烯衍生物8-11的合成a[0172][0173]a条件:(a)nahco3,i2,ki,h2o,0℃至室温,过夜;(b)naoh,etoh,0℃至室温,3h,然后氨溶液(28-30%),etoh,45℃,过夜;(c)4-茴香醛,acoh,nabh(oac)3,dce,75℃,过夜;(d)xtalfluor-m,(hf)3et3n,dcm,在塑料容器中,室温,过夜;(e)pd(oh)2,boc2o,meoh,etoac,室温,过夜;(f)phsecl,khmds(1m,在thf中),-78℃;(g)m-cpba,dcm,室温,2h;(h)hcl(4m),acoh,80℃,过夜;(i)boc2o,etoh,0℃至室温,5h;(j)pcc,dcm,室温,过夜。
[0174]
氟取代的环己烯类似物的动力学研究。体外研究表明,氟取代的环己烯类似物8-11抑制hoat,具有好的选择性(相对于gaba-at)(表1)。结果表明,6元环类似物对gaba-at的结合亲和力低于1,具有大得多的ki值,表明较大的环尺寸可能干扰类似物与gaba-at之间的结合相互作用,所述gaba-at具有相对小且刚性的活性部位。选择性与我们的初始设计策略和初始结合步骤的对接结果一致
[0175]
(数据未显示)。
[0176]
表1.化合物1、7-11和(s)-氨己烯酸对hoat和gaba-at的钝化的动力学常数a[0177][0178]
a k
inact
和ki值通过以下方程式来确定:k
obs
=k
inact
*[i]/(ki+[i]),并表示为平均值和标准误差。
b k
inact
/ki的比率通过k
obs
=k
inact
*[i]/(ki+[i])的斜率来确定。k
inact
大于最大k
obs
,后者在时间依赖性测定中确定;ki大于k
obs
(最大)/比率。c在ph 6.5试验的测定。d参考文献40。
9,在空间群p3112中,还发现一个不对称单元含有3个单体。可以通过晶体学对称性观察生物组装体。将hoat-8和hoat-9的最终模型修正(refine)至2.20和的分辨率,rfree/rwork值分别为15.70%/19.90%和27.49%/23.98%。最终的修正统计显示在实施例2、表3中。通过省略plp、配体和lys292生成每个结构的polder图
42
(数据未显示)。plp的密度在两种结构的polder图中都清楚地表示出来。还观察了8和9的密度,清楚地表明环和羧酸根部分的存在,正如关于最终加合物所预期的那样。hoat-8/9的解析结构已存入pdb库(pdb代码:6v8d和6v8c)。
[0188]
在hoat-8和hoat-9的活性部位内均观察到三元加合物。基于钝化的hoat-8和hoat-9的活性部位对比,8和9与plp和lys292反应并共价连接,尽管在不同的位置,但各自形成三元加合物。在hoat-8晶体结构中,plp和lys292与钝化剂连接在中心碳原子处,得到16a。该结果与ms数据一致,并排除了22作为最终加合物,其中plp和lys292与钝化剂在不同位置形成共价键。因此,途径a是8的最可能的钝化机制,而不是途径c(方案2)。在hoat-9晶体结构中,lys292和plp与钝化剂形成共价键以产生26。没有观察到密度表明氟原子留在加合物上。氟的完全丢失和加合物上共价键的存在因此排除了途径b(方案2)作为钝化的可能机制,如通过完整蛋白质谱法所得出的结论。在hoat-9的活性部位中的三元加合物在环上的两个单独的位置连接,而不是如在17中在中心原子处。polder图(fo-fc)产生的电子密度完全包围了在赖氨酸和plp之间形成的c-n键,表明9通过途径c将hoat钝化为26(方案2)。此外,thr322位于6元环的中心,并似乎与加合物形成孤对-芳族相互作用(lone-pair-aromatic interaction)
43
(数据未显示),这支持了26的形成。
[0189]
如在图3(右)中所示,26的羧酸根上的两个氧原子都与tyr55建立氢键(2.7和),而16a的羧酸根上的仅一个氧原子(图3,左)被观察到参与与tyr55的氢键与tyr55的相互作用具有优先性;在钝化的hoat-7的晶体结构中存在类似的相互作用
31
。观察到16a和26的羧酸根与gly320的肽主链形成n-π*相互作用(二者都是),但与arg180没有相互作用,这对于天然底物识别至关重要
43
。与gaba-at的活性位点相比,tyr55是hoat的活性部位中的一个独特残基,其被认为与鸟氨酸的α-氨基形成氢键
44,45
。gaba类似物8和9与tyr55的相互作用有助于它们的高效力,即使它们比天然底物鸟氨酸少一个碳。
[0190]
转换机制。此前,发现1可以通过氨基转移酶转化为代谢物,并释放plp或pmp
27
。考虑到1和8/9之间的结构相似性,提出了两种可能的转换途径,如在方案4中所示。plp与8/9的希夫碱形成和随后的赖氨酸辅助的去质子化产生中间体13a/13b。中间体13a/13b可以通过辅酶的直接质子化转化为中间体37a/37b。形成的亚胺可以进一步水解,并释放pmp和代谢物38a/38b。该pmp转换机制类似于hoat对鸟氨酸的降解。中间体13a/13b也可以经历氟离子的消除和亚胺的水解,并释放烯胺15a/15b,它们可以水解得到酮39a/39b。在该plp转换机制中,即使在没有α-kg存在下,再生的plp也会进一步与钝化剂形成希夫碱。为了确定在8和9对hoat的钝化过程中是否涉及上述转换机制中的任何一种,我们对这两种钝化剂进行了分配比实验、氟离子释放测定和代谢物质谱分析。
[0191]
方案4.提出的hoat对8或9的转换机制
[0192][0193]
分配比和氟释放。分配比是充当底物的化合物与使酶钝化的化合物的比率。在理想情况下,剩余的酶活性vs添加的化合物当量的图将给出从100%到0%剩余酶活性的直线。与x轴的截距给出了钝化每个酶分子所需的钝化剂分子的数目(转换数)。这个数目包括钝化酶所需的一分子钝化剂;因此,分配比是转换数减一(假定钝化剂与酶的化学计量比为1:1)。因此,通过用不同当量的钝化剂和已知量的hoat滴定酶,确定8/9的分配比(图9和10)。外推线性关系以产生完全钝化酶所需的精确当量。由此,我们确定8的分配比为22.0和9的分配比为0.8。
[0194]
在plp和pmp转换途径中可以释放不同当量的氟离子
29,31
。在没有α-kg存在下,如果在转换机制中形成pmp,则它不可被转化回plp,这导致少于一当量的释放的氟离子。如果在释放氟离子后在转换机制中再生plp,则会释放多个当量的氟离子。使用氟离子选择电极,
可以检测每次酶转换释放的氟离子的数目。当在没有α-kg存在下用过量的8钝化hoat时,每个酶活性部位释放22.7当量的氟离子(实施例2,表4)。在9的情况下,每个酶活性部位释放2.9当量的氟离子(实施例2,表4)。这些结果指示,8和9的转换机制必须再生plp作为转换机制的一部分,并在钝化前释放额外当量的氟离子,这分别与它们的分配比一致。
[0195]
基于质谱法的代谢物分析。在钝化的hoat样品8/9的尺寸排阻过滤后,通过非靶向代谢组学(
±
esi hrms)分析滤液以检测代谢物。检测在plp转换途径中的代谢物39a(m/z 139.0393,[m-h]-1)和39b(m/z 157.0301,[m-h]-1),并通过其碎裂波谱确认(数据未显示)。通过hrms监测plp和pmp的标准品,并将基于hcd的前体碎片用于确认代谢物检测和保留时间(数据未显示)。在8和9的样品中,通过质谱法仅检测到plp(m/z 248.0316,[m+h]+1);既没有观察到pmp,也没有观察到pmp的加合物(数据未显示)。总之,代谢组学结果表明这两种化合物经历相似的plp再生转换机制(方案4)。
[0196]
似乎合理的8和9与hoat的机制。基于上述钝化和转换机制研究,钝化剂8的改进机制途径显示在方案5中。最初形成希夫碱12a,并随后去质子化以提供中间体13a。基于氟离子释放实验和代谢组学结果,中间体13a完全转化为中间体14a。然后希夫碱14a在plp亚胺位置而不是共轭烯烃位置被lys 292攻击。根据8的分配比,只有4%的活性烯胺15a攻击lys-plp复合物以进行钝化(途径a),且其中96%水解以产生39a(途径b)。
[0197]
方案5.似乎合理的8与hoat的机制
[0198][0199]
方案6.似乎合理的9与hoat的机制
[0200][0201]
如在方案6中所示,提出了钝化剂9的改进机制途径。尽管通过类似的氟离子释放过程形成活性中间体14b,但通过在共轭双键处的赖氨酸加成发生钝化。形成的加合物进一步转化为更稳定的芳族加合物(26;56%,途径a),且44%的中间体14b转化为惰性烯胺15b,其不攻击lys-plp复合物,反而是被水解以产生代谢物39b(途径b)。
[0202]
通过完整蛋白ms及其晶体结构复合物确认加合物16a/26的结构,并通过质谱测定法检测到对应的代谢物39a/39b,同时释放多个当量的氟离子。总之,单个氟原子的差异导致8(烯胺途径)和9(加成-芳构化途径)的不同钝化机制途径,但通过相同的转换机制(plp途径)。
[0203]
分子动力学(md)模拟和静电势(esp)电荷计算。尽管在8和9的钝化过程中形成了非常相似的中间体(14a/14b和15a/15b),但所得的共价加合物(分别为16a和26)却截然不同。在以前,丙氨酸消旋酶抑制剂的不同钝化机制被证明是不同数目的氟(一个氟vs三个氟)的结果
46
。为了更好地理解额外的氟原子对这些中间体的影响,进行了分子对接/动力学和esp电荷计算以证明它们在hoat的催化口袋中的结合位姿以及与lys292或lys292-plp复合物的反应亲和力。
[0204]
基于分子对接研究,14a和14b在hoat的活性部位中的对接位姿是几乎相同的,其中lys292位于两个潜在亲电子位点的中间:plp亚胺部分的碳(c4′
)和共轭烯烃的末端碳(c
δ
)(数据未显示)。进行分子动力学模拟以分别计算亲核lys292的氮(n
lys
)与14a/14b的
c4′
/c
δ
位置之间的平均距离(数据未显示)。在14a中的n
lys-c4′
和n
lys-c
δ
的平均距离是非常相似的,而14b的c4′
比14b的c
δ
更靠近n
lys
,平均相差在分子动力学模拟期间还监测了环己二烯环和吡啶环之间的平均双面夹角(数据未显示)。14b的平均双面夹角(-148.26
°
)比14a的平均双面夹角(-91.64
°
;正交且非共轭)更接近-180
°
(完全共轭),表明14b中的烯烃具有更大的与吡啶环的共轭倾向。计算了中间体14a/b和15a/b的esp电荷和电子密度/静电势图以评价额外氟原子的影响(数据未显示)。由于额外氟原子的引入,在c
δ-14a(-0.28)和c
δ-14b(+0.49)之间表现出显著差异,在c
δ-15a(-0.70)和c
δ-15b(+0.07)之间也是如此,而在c4′-14a处的电荷(+0.42)接近于在c4′-14b处的电荷(+0.45)。因此,从分子动力学模拟(数据未显示)来看,lys292向c
δ-14a和c4′-14a的可达性是相似的,但双面夹角(-91.64
°
)和碳电荷(-0.28vs+0.42)明显不利于lys292对14a的烯烃的亲核攻击;在c4′
处的攻击产生烯胺15a并导致最终的加合物16a(方案5)。在14b的情况下,距离n
lys-c4′
有利于lys在14b的c
4'
处的亲核攻击(相对于14b的c
δ
)(vs),但额外的氟原子大大增强了14b的c
δ
的亲电子性,导致最终加合物26和无活性烯胺15b的产生,后者被质子化并被水解为39b(方案6)。在15b中的额外的氟原子显著降低了其亲核性,从而阻止了加合物16b和17的形成(方案2)。分子对接/动力学和esp电荷计算都清楚地合理化了我们在8和9对hoat的钝化期间的观察结果。
[0205]
结论
[0206]
在过去的几年中,对人鸟氨酸氨基转移酶(hoat)的选择性抑制已逐渐被认为是癌症、尤其是肝细胞癌(hcc)的潜在治疗
11
。基于非选择性钝化剂1的已知钝化机制
31
,借助于分子动力学模拟和对接,合理设计了一类新型氟取代的环己烯类似物(8-11),并进行合成和评价。在它们中,与1相比,类似物8和9表现出显著提高的对hoat的抑制活性,具有优秀的选择性(相对于gaba-at)(表1)。借助于透析实验、总转换和氟离子释放测量,通过质谱法和晶体学阐明了8和9的钝化途径。尽管单氟取代的类似物8通过烯胺途径钝化hoat(方案2,途径a),但二氟类似物9通过新型加成芳构化机制钝化hoat(方案2,途径c),这有助于其显著增强的效力。值得注意的是,以小于1da的误差获得了非常准确的完整蛋白质量,从而允许在其晶体结构之前轻松确定加合物质量(表2)。令人感兴趣的是,尽管钝化机制不同,但hoat对8和9的转换机制几乎相同(方案4,plp途径),并借助于靶向质谱法鉴定代谢物。通过使用分子动力学(md)模拟和静电势(esp)电荷计算来支持和合理解释8和9的似乎合理的钝化和转换机制,表明分子中的单个氟原子差异可以控制酶钝化机制。
[0207]
参考文献
[0208]
(1)sayiner,m.;golabi,p.;younossi,z.m.disease burden of hepatocellular carcinoma:a global perspective.dig.dis.sci.2019,64,910-917.
[0209]
(2)personeni,n.;rimassa,l.hepatocellular carcinoma:a global disease in need of individualized treatment strategies.j.oncol.pract.2017,13,368-370.
[0210]
(3)sherman,m.;bruix,j.;porayko,m.;tran,t.;comm,a.p.g.screening for hepatocellular carcinoma:the rationale for the american association for the study of liver diseases recommendations.hepatology 2012,56,793-796.
[0211]
(4)yang,j.d.;roberts,l.r.hepatocellular carcinoma:a global view.nat.
axis and cancer.front.oncol.2012,2,60.
[0225]
(18)tang,l.;zeng,j.;geng,p.y.;fang,c.n.;wang,y.;sun,m.j.;wang,c.s.;wang,j.;yin,p.y.;hu,c.x.;guo,l.;yu,j.e.;gao,p.;li,e.y.;zhuang,z.p.;xu,g.w.;liu,y.global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular carcinoma.clin.cancer res.2018,24,474-485.
[0226]
(19)heiden,m.g.v.;cantley,l.c.;thompson,c.b.understanding the warburg effect:the metabolic requirements of cell proliferation.science 2009,324,1029-1033.
[0227]
(20)colnot,s.;decaens,t.;niwa-kawakita,m.;godard,c.;hamard,g.;kahn,a.;giovannini,m.;perret,c.liver-targeted disruption of apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas.proc.natl.acad.sci.u.s.a.2004,101,17216-17221.
[0228]
(21)cadoret,a.;ovejero,c.;terris,b.;souil,e.;levy,l.;lamers,w.h.;kitajewski,j.;kahn,a.;perret,c.new targets of beta-catenin signaling in the liver are involved in the glutamine metabolism.oncogene 2002,21,8293-8301.
[0229]
(22)liu,y.f.;wu,l.;li,k.;liu,f.r.;wang,l.;zhang,d.l.;zhou,j.;ma,x.;wang,s.y.;yang,s.y.ornithine aminotransferase promoted the proliferation and metastasis of non-small cell lung cancer via upregulation of mir-21.j.cell.physiol.2019,234,12828-12838.
[0230]
(23)markova,m.;peneff,c.;hewlins,m.j.e.;schirmer,t.;john,r.a.determinants of substrate specificity in omega-aminotransferases.j.biol.chem.2005,280,36409-36416.
[0231]
(24)mehta,p.k.;hale,t.i.;christen,p.aminotransferases-demonstration of homology and division into evolutionary subgroups.eur.j.biochem.1993,214,549-561.
[0232]
(25)silverman,r.b.design and mechanism of gaba aminotransferase inactivators.treatments for epilepsies and addictions.chem.rev.2018,118,4037-4070.
[0233]
(26)silverman,r.b.mechanism-based enzyme inactivators.methods enzymol.1995,249,240-283.
[0234]
(27)mascarenhas,r.;le,h.v.;clevenger,k.d.;lehrer,h.j.;ringe,d.;kelleher,n.l.;silverman,r.b.;liu,d.selective targeting by a mechanism-based inactivator against pyridoxal 5
′‑
phosphate-dependent enzymes:mechanisms of inactivation and alternative turnover.biochemistry 2017,56,4951-4961.
[0235]
(28)storici,p.;qiu,j.;schirmer,t.;silverman,r.b.mechanistic crystallography.mechanism of inactivation of gamma-aminobutyric acid aminotransferase by(1r,3s,4s)-3-amino-4-fluorocyclopentane-1-carboxylic acid as elucidated by crystallography.biochemistry 2004,43,14057-14063.
crystalline deoxofluorinating reagents.org.lett.2009,11,5050-5053.
[0247]
(40)pan,y.;qiu,j.;silverman,r.b.design,synthesis,and biological activity of a difluoro-substituted,conformationally rigid vigabatrin analogue as a potent gamma-aminobutyric acid aminotransferase inhibitor.j.med.chem.2003,46,5292-5293.
[0248]
(41)lee,h.;doud,e.h.;wu,r.;sanishvili,r.;juncosa,j.i.;liu,d.l.;kelleher,n.l.;silverman,r.b.mechanism of inactivation of gamma-aminobutyric acid aminotransferase by(1s,3s)-3-amino-4-difluoromethylene-1-cyclopentanoic acid(cpp-115).j.am.chem.soc.2015,137,2628-2640.
[0249]
(42)liebschner,d.;afonine,p.v.;moriarty,n.w.;poon,b.k.;sobolev,o.v.;terwilliger,t.c.;adams,p.d.polder maps:improving omit maps by excluding bulk solvent.acta.crystallogr.d 2017,73,148-157.
[0250]
(43)egli,m.;sarkhel,s.lone pair-aromatic interactions:to stabilize or not to stabilize.acc.chem.res.2007,40,197-205..
[0251]
(44)montioli,r.;paiardini,a.;giardina,g.;zanzoni,s.;cutruzzola,f.;cellini,b.;voltattorni,c.b.r180t variant of delta-ornithine aminotransferase associated with gyrate atrophy:biochemical,computational,x-ray and nmr studies provide insight into its catalytic features.febs j.2019,286,2787-2798.
[0252]
(45)storici,p.;capitani,g.;muller,r.;schirmer,t.;jansonius,j.n.crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine.j.mol.biol.1999,285,297-309.
[0253]
(46)faraci,w.s.;walsh,c.t.mechanism of inactivation of alanine racemase byβ,β,β-trifluoroalanine.biochemistry 1989,28,431-437.
[0254]
实施例2-实施例1的补充材料
[0255]
一般合成方法.
[0256]
所有化学物质购自sigma aldrich、acros organics或combiblock,并不经进一步纯化地使用。将无水溶剂(thf)在使用前通过穿过由活性氧化铝和负载型铜氧化还原催化剂组成的柱进行纯化。收率是指色谱上均质的材料。使用merck silica gelf-254预制板(0.25mm厚度)进行分析型薄层色谱法(tlc),并通过紫外线(254nm)和/或钼酸铵铈染色和/或茚三酮染色使组分可视化。在具有各种taledyne筒(4-80g,40-63μm,)的teledyne combiflash rf加自动化快速纯化系统上进行快速柱色谱法。除非另外指出,否则用己烷类和乙酸乙酯进行纯化。在bruker avance-iii nmr谱仪上在cdcl3、cd3od或dmso-d6中分别在500mhz和126mhz记录1h和13c nmr谱。以ppm为单位报告化学位移;如下指示多重性:s=单峰,d=双峰,t=三重峰,q=四重峰,dd=双二重峰,dt=双三重峰,dq=双四重峰,m=多重峰共振。以hz为单位报告偶合常数
‘
j’。在西北大学综合分子结构教育和研究中心(integrated molecular structure education and research center(imserc),northwestern university),使用具有agilent g1312a hplc泵和agilent g1367b自动注
射器的电喷射电离,以阳离子模式在agilent 6210lc-tof波谱仪上获得高分辨率质谱图数据。使用具有phenomenex kintex c-18柱(50x 2.1mm,2.6μm)的反相agilent infinity 1260hplc进行分析型hplc,在254nm检测紫外吸光度。
[0257]
(1r,4r,5r)-4-碘-6-氧杂二环[3.2.1]辛烷-7-酮(28)。在冰浴中向nahco3(15.98g,190.74mmol,3.0当量)在水(200ml)中的搅拌溶液中缓慢地加入(r)-环己-3-烯-1-甲酸27(8g,63.41mmol,1.0当量)。逐份加入ki(52.36g,317.07mmol,5.0当量)和i2(17.7g,69.76mmol,1.1当量)。在30min以后除去冰浴并在室温搅拌过夜。将反应用饱和na2s2o3(150ml水溶液)淬灭,并用etoac(200ml)萃取3次。将合并的有机相用盐水(10ml)洗涤并用无水na2so4干燥。将溶液浓缩以产生棕色固体,将其分散在己烷(100ml)中并搅拌3h。将悬浮液过滤以产生淡棕色粉末(29,15.2g,95%)。1h nmr(500mhz,cdcl3)δ4.82(dd,j=5.9,4.2hz,1h),4.50(t,j=5.0hz,1h),2.79(d,j=12.3hz,1h),2.67(s,1h),2.48-2.35(m,2h),2.11(dd,j=16.5,5.3hz,1h),1.90(tdd,j=13.0,5.4,2.1hz,1h),1.82(dt,j=12.8,5.4hz,1h)。13c nmr(126mhz,cdcl3)δ177.9,80.4,38.7,34.6,29.9,23.9,23.3.lrms(apci)(m+h+):252.85。
[0258]
(1r,3s,4s)-3-(双(4-甲氧基苄基)氨基)-4-羟基环己烷-1-甲酸乙酯(29)。在冰浴中搅拌28(28.7g,118.9mmol,1.0当量)在etoh(100ml)中的悬浮液。逐滴加入naoh溶液(2m,65ml,130.8mmol,1.1当量)。加入以后除去冰浴,并将溶液在室温搅拌4h。将溶液在40℃以下浓缩,用水(100ml)稀释,并用dcm(200ml)萃取3次。将合并的有机相用水(100ml)和盐水(100ml)洗涤并用无水na2so4干燥。将溶液浓缩以产生棕色油(15.3g)。向得到的棕色油(10.5g)在etoh(50ml)中的溶液中加入氨水(28-30%,100ml)。将溶液加热至45℃并搅拌过夜。通过lc-ms确定反应结束。将溶液浓缩以产生粗制的氨基醇中间体(11.1g)。向粗制的中间体(6.0g,32.04mmol,1.0当量)在dce(200ml)中的溶液中加入4-茴香醛(13.1g,96.13mmol,3.0当量)和acoh(5.77g,96.13mmol,3.0当量)。将溶液加热至75℃并搅拌1h。向该溶液中历时2h逐份加入nabh(oac)3(20.37g,96.13mmol,3.0当量)。将得到的混合物在75℃搅拌过夜。通过lc-ms确定反应结束。将混合物冷却至室温并用dcm(200ml)和水(200ml)稀释。搅拌10min以后,将有机相分离,并然后依次用水(200ml)、nahco3(水溶液200ml)和盐水(100ml)洗涤。将有机相用无水na2so4干燥。将溶液浓缩并通过硅胶色谱法(50%etoac在己烷中)纯化以提供白色固体(29,6.297g,三步35%)。1h nmr(500mhz,cdcl3)δ7.17(d,j=8.5hz,4h),6.84(d,j=8.6hz,4h),4.17(dq,j=10.8,7.1hz,1h),4.07(dq,j=10.8,7.1hz,1h),3.79(s,1h),3.78(s,6h),3.74(d,j=13.1hz,2h),3.63(s,1h),3.47(td,j=10.4,4.5hz,1h),3.31(d,j=13.0hz,2h),2.80(dp,j=4.9,2.2hz,1h),2.53(ddd,j=12.7,9.8,3.4hz,1h),2.47(dq,j=12.9,2.7hz,1h),2.17(dp,j=14.0,3.1hz,1h),1.97(dq,j=11.6,3.4hz,1h),1.50-1.42(m,2h),1.23(t,j=7.1hz,3h)。13c nmr(126mhz,cdcl3)δ174.1,158.8,131.5,130.3,113.9,68.7,60.7,60.3,55.3,52.6,39.6,30.1,25.1,23.5,14.4.c
25h34
no5(m+h+)的hrms(esi)计算值:428.2431,实测:428.242。
[0259]
(1r,3s,4s)-3-(双(4-甲氧基苄基)氨基)-4-氟环己烷-1-甲酸乙酯(30)。在ar下在塑料容器中向xtalfluor-m(7.67g,31.65mmol,1.5当量)在dcm(100ml)中的搅拌悬浮液中缓慢地加入29(9.0g,21.05mmol,1.0当量)和(hf)3et3n(5.09g,31.65mmol,1.5当量)在dcm(50ml)中的溶液。加入结束后,将混合物在室温搅拌过夜。通过tlc(己烷:etoac=2:1)
确定反应结束。将反应用dcm(100ml)稀释并用水(50ml)淬灭。将有机相分离,并然后依次用水(100ml)、nahco3(水溶液100ml)和盐水(100ml)洗涤。将有机相用无水na2so4干燥。将溶液浓缩并通过硅胶色谱法(25%etoac在己烷中)纯化以提供无色油(30,6.81g,75%)。1h nmr(500mhz,cdcl3)δ7.30(d,j=8.5hz,4h),6.84(d,j=8.6hz,4h),4.62(dtd,j=50.5,10.3,4.8hz,1h),4.01(dq,j=10.8,7.1hz,1h),3.91(dq,j=10.8,7.1hz,1h),3.78(s,6h),3.73(d,j=13.6hz,2h),3.65(d,j=13.5hz,2h),2.85(dddd,j=12.3,9.7,8.4,3.9hz,1h),2.66(dh,j=5.5,2.8hz,1h),2.32(ddq,j=12.8,6.2,3.0hz,1h),2.19-2.10(m,1h),2.03(ddq,j=12.3,8.2,4.1hz,1h),1.64-1.47(m,2h),1.38(tddd,j=13.7,5.1,3.6,1.4hz,1h),1.08(t,j=7.1hz,3h)。13c nmr(126mhz,cdcl3)δ173.9,158.6,132.5,129.8,113.6,92.5(d,j=178.1hz),60.6,57.8(d,j=16.1hz),55.3,53.6,39.0(d,j=2.0hz),29.2(d,j=18.7hz),28.6(d,j=8.4hz),24.9(d,j=11.2hz),14.1.c
25h33
fno4(m+h+)的hrms(esi)计算值:430.2388,实测:430.238。
[0260]
(1r,3s,4s)-3-((叔-丁氧基羰基)氨基)-4-氟环己烷-1-甲酸乙酯(31)。在ar下向30(5.0g,17.46mmol,1.0当量)在meoh(50ml)和etoac(50ml)中的溶液中加入boc2o(3.81g,11.64mmol,1.0当量)和pd(oh)2(1.0g,20%wt)。将烧瓶抽真空以除去ar,并然后用h2气球重新填充。将混合物在室温搅拌过夜。通过lc-ms确定反应结束。将悬浮液穿过celite垫过滤,并用额外100ml的etoac洗涤。将滤液浓缩并通过硅胶色谱法(20%etoac在己烷中)纯化以提供白色固体(31,2.73g,81%)。1h nmr(500mhz,cdcl3)δ4.55(s,1h),4.47(d,j=39.7hz,1h),4.15(q,j=7.1hz,2h),3.89(s,1h),2.49(s,1h),2.23(ddt,j=13.3,8.5,4.1hz,1h),2.00-1.76(m,3h),1.76-1.64(m,2h),1.44(s,10h),1.26(t,j=7.1hz,3h).13c nmr(126mhz,cdcl3)δ174.3,155.2,90.589.8(d,j=173.2hz),60.8,49.3,38.0,29.8,26.7(d,j=20.2hz),23.4(d,j=5.4hz),14.4.19f nmr(564mhz,cd3od)δ-181.34(d,j=50.7hz)。c
14h24
fnnao4(m+na+)的hrms(esi)计算值:312.158,实测:312.1582。
[0261]
(4s,5s)-5-((叔-丁氧基羰基)氨基)-4-氟环己-1-烯-1-甲酸乙酯(32b)和(3s,4s)-3-((叔-丁氧基羰基)氨基)-4-氟环己-1-烯-1-甲酸乙酯(32a)。向khmds的搅拌溶液(1m在thf中,10.89ml,10.89mmol,2.1当量)中缓慢地加入干燥thf(10ml),并将31(1.5g,5.18mmol,1.0当量)在干燥thf(30ml)中的溶液历时30min在ar下在-78℃加入。将溶液在-78℃搅拌额外3h,随后加入phsecl(1.09mg,5.70mmol,1.1当量)在thf(10ml)中的溶液。然后将溶液缓慢地温热至室温并搅拌过夜。通过加入饱和nh4cl(20ml)淬灭反应。然后将溶液用etoac(200ml)稀释并将有机相分离。将水相用etoac(100ml)萃取2次。将合并的有机层用盐水(约30ml)洗涤并用无水na2so4干燥。将溶液浓缩并产生黄色油。向得到的油(1.55g,3.49mmol,1.0当量)在dcm(50ml)中的搅拌溶液中加入m-cpba(1.17g,5.23mmol,1.5当量)。将溶液在室温搅拌3h。通过lc-ms确定反应结束。将反应用na2s2o3(水溶液10ml)淬灭。将有机相分离,用饱和nahco3(水溶液10ml)和盐水(10ml)洗涤,并用无水na2so4干燥。将溶液浓缩并通过硅胶色谱法(5-10%etoac在己烷中)和c-18色谱法纯化以提供白色固体(32b,92mg,9%),1h nmr(500mhz,cdcl3)δ6.85(tq,j=3.5,1.9hz,1h),4.75(dq,j=47.5,4.9,4.2hz,1h),4.52(s,1h),4.20(q,j=7.1hz,2h),4.07(s,1h),2.83(d,j=17.9hz,1h),2.66(t,j=23.7hz,1h),2.53(t,j=19.4hz,1h),2.33(ddq,j=18.1,5.5,1.9hz,1h),1.44(s,9h),1.29(t,j=7.1hz,3h).13c nmr(126mhz,cdcl3)δ166.1,155.3,134.5(d,j=4.2hz),
128.0,87.1(d,j=176.2hz),60.8,47.7,29.5(d,j=22.6hz),28.3,27.6(d,j=3.2hz),14.2.c
14h22
fnnao4(m+na+)的hrms(esi)计算值:310.1425,实测:310.1416。
[0262]
还得到另一份白色固体(32a,365mg,36%)。1h nmr(500mhz,dmso-d6)δ7.27(d,j=8.4hz,1h),6.46(s,1h),4.60(dddd,j=49.1,9.3,5.9,3.1hz,1h),4.25(ddq,j=14.5,9.0,3.4,2.9hz,1h),4.13(q,j=7.1hz,2h),2.38-2.20(m,2h),2.05-1.94(m,1h),1.88(dp,j=14.1,6.9hz,1h),1.40(s,9h),1.22(t,j=7.1hz,3h).13c nmr(126mhz,dmso-d6)δ165.5,155.1,136.2,136.2,130.7,90.0(d,j=175.2hz),78.4,60.3,50.4(d,j=25.0hz),28.1,25.3(d,j=19.4hz),21.5(d,j=9.5hz),14.1.c
14h22
fnnao4(m+na+)的hrms(esi)计算值:310.1425,实测:310.1416.;和未分离的混合物(421mg,42%)。
[0263]
(1r,3s)-3-((叔-丁氧基羰基)氨基)-4-氧代环己烷-1-甲酸乙酯(33)。将28(16g,63.41mmol)分散在etoh(100ml)中形成悬浮液并在冰浴中搅拌。逐滴加入naoh溶液(2m,35ml,69.76mmol,1.1当量)。加入以后除去冰浴,并将溶液在室温搅拌4h。将溶液在40℃以下浓缩,用水(100ml)稀释,并用dcm(200ml)萃取3次。将合并的有机相用水(100ml)和盐水(100ml)洗涤并用无水na2so4干燥。将溶液浓缩并用etoh(20ml)和氨水溶液(28-30%,40ml)稀释。将溶液加热至45℃并搅拌过夜。通过lc-ms确定反应结束。将溶液浓缩并用etoh(50ml)稀释。将溶液浓缩,用etoh(200ml)稀释,并在冰浴中搅拌。逐份加入boc2o(13.84g,63.41mmol,1.0当量)。然后将溶液温热至室温并搅拌4h。将溶液浓缩以产生粗制的33(纯化用于结构确定:1h nmr(500mhz,cdcl3)δ4.61(d,j=7.0hz,1h),4.19-4.12(m,2h),3.57(s,1h),3.47-3.28(m,2h),2.62(p,j=4.5hz,1h),2.35-2.26(m,1h),2.15-2.07(m,1h),1.90-1.84(m,1h),1.55-1.48(m,2h),1.44(s,10h),1.25(q,j=7.0hz,3h).13c nmr(126mhz,cdcl3)δ174.2,171.3,80.2,73.8,60.8,53.0,38.9,31.4,30.1,28.5,24.8,14.4.)。c
14h25
n nao5(m+na+)的hrms(esi)计算值:310.1625,实测:310.1618。
[0264]
将粗制的33溶解在dcm(300ml)中并在冰浴中搅拌。逐份加入pcc(27.34g,126.83mmol,2.0当量)。将混合物温热至室温并搅拌过夜。通过tlc(己烷:etoac=2:1)确定反应结束。将混合物穿过薄硅胶垫过滤,并用dcm(50ml)洗脱3次。将溶液浓缩并通过硅胶色谱法(40%etoac在己烷中)纯化以提供白色固体(34,10.2g,56%)。1h nmr(400mhz,cdcl3)δ5.36(s,1h),4.48(s,1h),4.25(q,j=6.7hz,2h),2.99-2.91(m,1h),2.87(s,1h),2.70(td,j=13.7,5.7hz,1h),2.46(t,j=14.1hz,2h),1.87(tt,j=14.3,4.8hz,1h),1.62(td,j=12.8,4.9hz,1h),1.43(s,9h),1.32(t,j=7.1hz,3h)。c
14h23
nnao5(m+na+)的hrms(esi)计算值:308.1468,实测:308.1459。
[0265]
(1r,3s)-3-((叔-丁氧基羰基)氨基)-4,4-二氟环己烷-1-甲酸乙酯(35)。在ar下在塑料容器中向xtalfluor-m(2.04g,8.41mmol,2.0当量)在dcm(20ml)中的搅拌悬浮液中缓慢地加入34(1.2g,4.21mmol,1.0当量)和(hf)3et3n(1.36g,8.41mmol,2.0当量)在dcm(10ml)中的溶液。加入结束后,将混合物在室温搅拌过夜。通过tlc(己烷:etoac=2:1)确定反应结束。将反应用dcm(30ml)稀释并用水(10ml)淬灭。将有机相分离,并然后用水(10ml)、nahco3(水溶液10ml)和盐水(10ml)洗涤。将有机相用无水na2so4干燥。将溶液浓缩并通过硅胶色谱法(20%etoac在己烷中)纯化以提供白色固体(35,629mg,48%)。1h nmr(500mhz,cdcl3)δ4.68(s,1h),4.18(q,j=7.1hz,2h),4.12(s,1h),2.71(s,1h),2.31(d,j=12.3hz,1h),2.14-1.90(m,3h),1.81-1.72(m,1h),1.71-1.65(m,1h),1.45(s,9h),1.28(t,j=
7.1hz,3h).13c nmr(126mhz,cdcl3)δ13c nmr(126mhz,cdcl3)δ173.5,155.2,121.6(dd,j=248.2,242.0hz),80.1,61.1,50.1(dd,j=25.0,21.0hz),37.9,31.5,30.4(t,j=23.3hz),28.4,23.8(d,j=7.9hz),14.4.c
14h23
f2nnao4(m+na+)的hrms(esi)计算值:330.q1487,实测:330.1479。
[0266]
(s)-3-((叔-丁氧基羰基)氨基)-4,4-二氟环己-1-烯-1-甲酸乙酯(36a)和(s)-5-((叔-丁氧基羰基)氨基)-4,4-二氟环己-1-烯-1-甲酸乙酯(36b)。将khmds的搅拌溶液(1m在thf中,7.16ml,7.16mmol,2.2当量)在干燥thf(5ml)中稀释并在ar下冷却至-78℃;然后历时30min缓慢地加入35(1.0g,3.25mmol,1.0当量)在干燥thf(10ml)中的溶液。将溶液在-78℃搅拌额外3h,随后加入phsecl(685mg,3.58mmol,1.1当量)在thf(10ml)中的溶液。然后将溶液缓慢地温热至室温并搅拌过夜。通过加入饱和nh4cl(20ml)淬灭反应。然后将溶液用etoac(200ml)稀释,并将有机相分离。将水相用etoac(100ml)萃取2次。将合并的有机相用盐水(约30ml)洗涤并用无水na2so4干燥。将溶液浓缩以产生黄色油。向油在dcm(20ml)中的搅拌溶液中加入m-cpba(555mg,2.48mmol,1.5当量)。将溶液在室温搅拌3h。通过lc-ms确定反应结束。将反应用na2s2o3(水溶液10ml)淬灭。将有机相分离,用饱和nahco3(水溶液10ml)和盐水(10ml)洗涤,并用无水na2so4干燥。将溶液浓缩并通过硅胶色谱法(5-10%etoac在己烷中)纯化以提供白色固体(36a,258mg,51%),1h nmr(400mhz,cdcl3)δ6.59(ddt,j=5.2,2.6,1.3hz,1h),4.90-4.i68(m,2h),4.21(q,j=7.1hz,2h),2.70-2.57(m,1h),2.57-2.43(m,1h),2.28(tdd,j=16.6,8.6,4.5hz,1h),2.17-1.97(m,1h),1.47(s,9h),1.30(t,j=7.1hz,3h).13c nmr(126mhz,cdcl3)δ165.8,155.4,135.6,131.9,120.2(dd,j=247.1,241.3hz),80.7,61.2,51.4(t,j=26.8,22.4hz),29.5(t,j=23.5hz),28.4,22.8(dd,j=7.6,2.9hz),14.3.19f nmr(376mhz,cdcl3)δ-105.13(d,j=239.4hz),-115.81(d,j=239.3hz),c
14h21
f2nnao4(m+na+)的hrms(esi)计算值:328.1331,实测:328.1324。
[0267]
还得到另一份白色固体(36b,62mg,12%)。1h nmr(500mhz,cdcl3)δ6.76(s,1h),4.83-4.67(m,1h),4.21(q,j=7.1hz,2h),4.18-4.14(m,1h),2.95(d,j=17.5hz,1h),2.90-2.70(m,2h),2.40-2.28(m,1h),1.46(s,9h),1.29(t,j=7.1hz,3h).13c nmr(126mhz,cdcl3)δ165.6,155.4,132.8(d,j=9.1hz),129.0,120.5(dd,j=246.9,242.0hz),80.4,61.1,49.5(t,j=21.5hz),34.8(t,j=28.1hz),30.3(t,j=3.6hz),28.4,14.3.,c
14h21
f2nnao4(m+na+)的hrms(esi)计算值:328.1331,实测:328.1324.;和未分离的36a和36b的混合物(94mg,19%)。
[0268]
一般去保护程序a。在ar下向hcl水溶液(4m,1.5ml)和acoh(1.5ml)的溶液中加入酯中间体。将溶液密封并加热至80℃并搅拌过夜。通过lc-ms确定反应结束。将溶液浓缩并通过c-18色谱法纯化以产生产物。
[0269]
(3s,4s)-3-氨基-4-氟环己-1-烯-1-甲酸盐酸盐(8)。将32a(150mg,0.522mmol)通过程序a去保护以产生白色固体(8,94mg,92%)。1h nmr(500mhz,cd3od)δ6.67(dt,j=5.1,2.5hz,1h),4.78(dddd,j=50.5,11.6,7.8,4.0hz,1h),4.27-4.13(m,1h),2.65(d,j=20.3hz,1h),2.50-2.39(m,1h),2.33-2.23(m,1h),1.98(ddq,j=17.5,11.9,6.0hz,1h).13c nmr(126mhz,cd3od)δ168.2,137.2(d,j=2.4hz),130.5(d,j=7.0hz),90.9(d,j=177.6hz),53.6(d,j=23.2hz),27.3(d,j=18.7hz),24.4(d,j=11.2hz).13c nmr(126mhz,cd3od)δ168.4,136.3(d,j=11.1hz),128.4,89.8(d,j=177.5hz),51.7(d,j=
18.1hz),31.8(d,j=21.7hz),29.2(d,j=6.1hz)。c7h
11
fno2(m+h+)的hrms(esi)计算值:160.0768,实测:160.0765。
[0270]
(s)-3-氨基-4,4-二氟环己-1-烯-1-甲酸盐酸盐(9)。将36a(105mg,0.34mmol)通过程序a去保护以产生白色固体(9,56mg,76%)。1h nmr(500mhz,cd3od)δ6.72(s,1h),4.65-4.52(m,1h),2.71(ddd,j=18.9,6.2,3.0hz,1h),2.55(dtd,j=19.0,6.4,3.1hz,1h),2.47-2.22(m,2h).13c nmr(126mhz,cd3od)δ167.9,136.8,129.62(d,j=5.9hz),121.05(t,j=246.2hz),52.27(dd,j=29.1,22.2hz),29.24(t,j=22.7hz),23.76(dd,j=7.7,2.9hz).19f nmr(376mhz,cd3od)δ-108.4(dp,j=240.6,5.8,5.3hz),-114.2(dq,j=240.7,23.2hz)。c7h
10
f2no2(m+h+)的hrms(esi)计算值:178.0674,实测:178.0672。
[0271]
(4s,5s)-5-氨基-4-氟环己-1-烯-1-甲酸盐酸盐(10)。将32b(80mg,0.278mmol)通过程序a去保护以产生白色固体(10,49mg,90%)。1h nmr(500mhz,cd3od)δ6.89(s,1h),5.02-4.95(m,1h),3.65(tt,j=10.6,6.2hz,1h),3.03(dt,j=17.3,5.8hz,1h),2.93(d,j=18.5hz,1h),2.65-2.51(m,1h),2.51-2.38(m,1h).13c nmr(126mhz,cd3od)δ168.4,136.3(d,j=11.1hz),128.4,89.8(d,j=177.5hz),51.7(d,j=18.1hz),31.8(d,j=21.7hz),29.2(d,j=6.1hz)。c7h
11
fno2(m+h+)的hrms(esi)计算值:160.0768,实测:160.0762。
[0272]
(s)-5-氨基-4,4-二氟环己-1-烯-1-甲酸盐酸盐(11)。将36b(30mg,0.10mmol)通过程序a去保护以产生白色固体(11,14mg,66%)。1h nmr(500mhz,cd3od)δ6.87(p,j=3.9hz,1h),4.06-3.95(m,1h),3.15-3.06(m,1h),3.00(dp,j=21.4,4.1,3.2hz,2h),2.53(ddd,j=17.1,10.6,2.8hz,1h).13c nmr(126mhz,cd3od)δ167.9,134.4(d,j=10.3hz),128.3,121.1(dd,j=245.6,243.5hz),50.9(t,j=22.5hz),34.9(t,j=25.6hz),28.6。c7h
10
f2no2(m+h+)的hrms(esi)计算值:178.0674,实测:178.0675。
[0273]
对接模拟
[0274]
使用分子操作环境(molecular operating environment,moe)计算套件的builder实用程序开发了与gaba-at或oat结合的配体的对接模型
1-3
。使用力场mmff94x在气相中进行配体的能量最小化,然后进行构象检索(conformational search)方案以生成结构-构象数据库。将天然gaba-at(pdb:1ohv)、天然hoat(1oat)、钝化的gaba-at(4y0i)和钝化的hoat(1gbn)的x-射线晶体结构分别上载至moe,随后进行受体制备。将在5vwo和4y0i的活性袋中的紧密结合的产物删除,并将催化性lys中和。将对接位点指定在催化性lys原子处。在制备的氨基转移酶模型(其中不相关的底物和溶剂原子被钝化)中进行配体对接。配体放置采用具有affinity dg评分的alpha triangle方法,生成300个数据点,所述数据点使用具有gbvi/wsa dg评分的诱导契合方法进一步修正,以获得前50个对接结果。基于评分和报告的x射线结构,分析每种配体的对接结果以选择最佳对接位姿。然后在pymol中执行所有的绘制(renderings)。
[0275]
酶测定
[0276]
根据文献程序
4,5
,表达、生长和纯化hoat和pycr1。从猪脑中分离gaba-at并根据文献程序纯化6。根据之前的程序
7,8
,进行gaba-at、hoat、ala-at和asp-at的偶联酶测定。
[0277]
透析测定
[0278]
使用以前的方案
7,9,10
,进行透析实验。
[0279]
分配比实验
[0280]
使用以前的方案
7,9,10
,计算分配比。
[0281]
氟离子释放
[0282]
使用以前的方案7,进行氟离子释放测定。通过bsa测定和稀释度计算,确定样品中hoat的终浓度为16.9
±
0.04ug/ml(单体,0.37
±
0.01μm)。从不同浓度的naf(f,μm)产生电压的校正曲线(v,mv),以获得方程式:f-=10^((v-181.9)/-53.18)。为了准确检测氟离子浓度,将2.0μm的氟离子加入每个对照和样品。通过氟离子释放浓度和hoat浓度之比,计算每个活性部位释放的氟离子的数目。
[0283]
完整蛋白和小分子质谱法
[0284]
在amicon ultra 30kda分子量旋转过滤器(millipore)上用水将重组和处理的hoat脱盐10次。为了色谱分辨(resolve)hoat,使用dionex ultimate3000液相色谱法系统(thermo fisher)将0.5μg的hoat加载到3cm plrp-s(agilent)捕集柱上。用10%溶剂b(95%mecn/5%h2o/0.2%fa)和90%溶剂a(5%mecn/95%h2o/0.2%fa)的10-min等度梯度洗涤蛋白分析物。将hoat在内部制造的75μm id x 15cm长纳米孔毛细管柱上分辨,该柱填充了plrp-s树脂(agilent)。将lc系统以300nl/min的流速在以下梯度运行:0-10min10%溶剂b;10-12min至40%溶剂b;12-22min至90%溶剂b;22-24min在90%溶剂b;24-26min至10%溶剂b;26-30min在10%溶剂b等度。在orbitrap质量分析器中,在运行于低压蛋白模式的fusion lumos tribrid质谱仪(thermo fisher)上使用[m+24h+]+24默认电荷状态获取正电的完整谱esi数据。使用定制的纳米电喷雾电离源,静态喷雾电压为1700v。在500-2,000m/z窗口中收集数据,每个扫描事件平均20次微扫描,分辨率为7,500(200m/z),最大喷射时间为50ms,且自动增益控制(agc)的目标值为5e6电荷。手动对平均求和扫描进行反卷积以生成中性质量。如前所述,在q-exactive orbitrap质谱仪上通过高分辨率lc-ms/ms鉴定和表征小分子和代谢物质量9。
[0285]
hoat结晶
[0286]
晶体结构生长。将新鲜制备的酶(200μg)用ph 8.0的含有5mmα-酮戊二酸的50mm焦磷酸钾透析。加入化合物8/9(0.5mg),并将酶在用箔覆盖的小瓶中钝化12h。偶联酶测定指示没有活性。在hoat活性的完全钝化以后,将酶样品在50mm n-(羟甲基)甲基甘氨酸(tricine)ph 7.8中浓缩至6mg/ml的蛋白浓度。通过悬滴蒸汽扩散方法优化结晶,并如下设置:改变peg 1000(10-20%)、nacl(100-250mm)、甘油(20%-30%),将50mm n-(羟甲基)甲基甘氨酸ph 7.8保持恒定作为缓冲液。对于每个孔,设定1:1比例的孔滴:蛋白溶液。在含有16.5%peg 1000、240mm nacl、25%甘油的孔中生长具有最佳形态学和尺寸的晶体用于数据收集。将晶体转移至冷冻保护剂溶液(补充了30%甘油的孔溶液),然后在液氮中快速冷冻。
[0287]
数据收集和处理。在argonne national laboratory(anl)在ls-cat光束线21-id-d、advanced photon source(aps)收集单色数据。使用dectris eiger 9m检测器在100k在的波长收集衍射数据。使用hkl200011套件对数据集进行索引和集成。数据统计总结在表3中。
[0288]
表3.被8和9钝化的hoat的晶体结构的统计
[0289]
[0290][0291]
表4.8和9的氟离子释放的计算
[0292]
[0293]
模型构建和修正。通过使用phenix软件套件中的phaseer进行分子置换来解析hoat结构
12
。第一个搜索模型是基于先前公开的hoat结构(pdb代码:1oat)。该模型使用coot重建
13
,使用phenix进行修正,并在coot和uscf chimera中进行分析
14
。最终的修正统计数据报告在表3中。在uscf chimera中制作结构图。
[0294]
分子动力学模拟
[0295]
采用对接方案生成对应于用于经典md模拟的14a和14b的初始hoat复合物。为该研究选择的蛋白结构是钝化的hoat的x-射线晶体结构(pdb id:1gbn),分辨率为使用h++服务器确定氨基酸残基的质子化状态。使用gausian09软件在hf/6-31+(g)理论水平上优化14a和14b的结构
15
。使用mgltools软件包提供的autodocktools-1.5.6,进一步修正抑制剂和蛋白结构
16
。使用autodock4.2软件将修正的抑制剂结构对接到oat活性部位中。使用autogrid4.2软件生成以活性部位为中心的网格框,网格间距为沿x、y和z轴的尺寸为55
×
55
×
55点。将lamarckian遗传算法(ga)用于构象搜索,ga群体大小为150,最大评价数目为2 500 000。将100个生成的位姿根据它们的rmsd值(截止值)进行聚类,并选择最佳类的最低能量构象进行md模拟。在hf/6-31+g理论水平计算几何优化的抑制剂的静电势能(esp)(hf/6-31+g)。然后,将这些能量用于使用在amber12程序中可得到的前室模块中所采用的静电势平方拟合(esp)方法推导抑制剂的部分原子电荷
17
。这是用于推导原始amber力场的部分原子电荷的方法。用一般amber力场(gaff)处理抑制剂原子,并在推导参数之前手动调整自动分配的gaff原子类型以准确表示其化学环境。使用amber12程序的tleap和前室模块生成参数和拓扑文件。然后使用acpype将这些参数和拓扑文件转换为gromacs兼容格式
18
。以类似方式推导出共价结合的plp的力场参数和拓扑学。将具有共价结合的plp参数的修改后的amberff99sb-ildn力场用于模拟蛋白。使用在gromacs 5.1.2软件包中可得的pdb2gmx模块推导出oat二聚体的参数。使用gromacs 5.1.2软件进行所有的md模拟
19
。tip3p模型用作水模型。将蛋白-抑制剂复合物浸入装满tip3p水的十二面体盒中,其中边界从蛋白边缘向所有方向延伸至少1.8nm。然后,将nacl添加到系统中直至0.15m的浓度(生理nacl浓度)以中和系统的电荷。以最速下降法然后是共轭梯度法使溶剂化系统的能量最小化,直到它以不大于500kj mol-1
nm-1
的最大力收敛。
[0296]
得到的能量最小化周期系统是md模拟的起始配置,其在extreme science and engineering discovery environment(xsede)的帮助下进行
20
。在生产md模拟之前,将系统在两个步骤中进行平衡。首先,使它在恒定nvt(粒子数、体积和温度)集合中平衡1ns。然后将所得系统在恒定npt(粒子数、压强和温度)集合中平衡3ns。将温度和压强分别通过v-rescale恒温器(时间常数为0.4ps)和parrinello-rahmanbarostat(时间常数为2ps)控制在310k和1巴。在两个平衡步骤中,通过施加1000kj mol-1
nm-2
的力常数来限制重原子的位置。此后,将位置约束在两次运行(每次1ns)中从500kj mol-1
nm-2
逐渐降低到100kj mol-1
nm-2
。最后,在npt条件下进行平衡系统的生产md模拟15ns(时间步长为2fs),没有位置限制。在npt平衡期间,v-rescale恒温器被最准确的nos
é‑
hoover恒温器替代,并用于其余的模拟方案。用粒子网格ewald(pme)方法处理长程静电相互作用,而库仑和范德华相互作用在1.2nm处被切断。使用线性约束求解器(linear constraint solver,lincs)算法限制原子的键长。
[0297]
使用gromacs软件包中可用的距离、mindist和角度工具进行重要的距离和双面夹
角测量。
[0298]
静电势(esp)电荷计算
[0299]
在hf/6-31+g理论水平计算在气相中的几何优化的抑制剂(14a和14b)的静电势(esp)能量和电荷(hf/6-31+g)。将gaussain09程序的cubegen实用程序用于生成电子密度和静电势图,并使用vmd 1.9.2分子可视化程序进行可视化。
[0300]
参考文献
[0301]
1.heath,t.k.;lutz,m.r.;reidl,c.t.;guzman,e.r.;herbert,c.a.;nocek,b.p.;holz,r.c.;olsen,k.w.;ballicora,m.a.;becker,d.p.,practical spectrophotometric assay for the dape-encoded n-succinyl-l,l-diaminopimelic acid desuccinylase,a potential antibiotic target.plos one 2018,13.
[0302]
2.vilar,s.;cozza,g.;moro,s.,medicinal chemistry and the molecular operating environment(moe):application of qsar and molecular docking to drug discovery.curr.top.med.chem.2008,8,1555-1572.
[0303]
3.boyd,s.,molecular operating environment.chem.world 2005,2,66-66.
[0304]
4.christensen,e.m.;patel,s.m.;korasick,d.a.;campbell,a.c.;krause,k.l.;becker,d.f.;tanner,j.j.,resolving the cofactor-binding site in the proline biosynthetic enzyme human pyrroline-5-carboxylate reductase 1.j.biol.chem.2017,292,7233-7243.
[0305]
5.mascarenhas,r.;le,h.v.;clevenger,k.d.;lehrer,h.j.;ringe,d.;kelleher,n.l.;silverman,r.b.;liu,d.,selective targeting by a mechanism-based inactivator against pyridoxal 5'-phosphate-dependent enzymes:mechanisms of inactivation and alternative turnover.biochemistry 2017,56,4951-4961.
[0306]
6.churchich,j.e.;moses,u.,4-aminobutyrate aminotransferase-the presence of nonequivalent binding-sites.j.biol.chem.1981,256,1101-1104.
[0307]
7.lee,h.;doud,e.h.;wu,r.;sanishvili,r.;juncosa,j.i.;liu,d.l.;kelleher,n.l.;silverman,r.b.,mechanism of inactivation of gamma-aminobutyric acid aminotransferase by(1s,3s)-3-amino-4-difluoromethylene-1-cyclopentanoic acid(cpp-115).j.am.chem.soc.2015,137,2628-2640.
[0308]
8.juncosa,j.i.;lee,h.;silverman,r.b.,two continuous coupled assays for ornithine-delta-aminotransferase.anal.biochem.2013,440,145-149.
[0309]
9.moschitto,m.j.;doubleday,p.f.;catlin,d.s.;kelleher,n.l.;liu,d.;silverman,r.b.,mechanism of inactivation of ornithine aminotransferase by(1s,3s)-3-amino-4-(hexafluoropropan-2-ylidenyl)cyclopentane-1-carboxylic acid.j.am.chem.soc.2019,141,10711-10721.
[0310]
10.juncosa,j.i.;takaya,k.;le,h.v.;moschitto,m.j.;weerawarna,p.m.;mascarenhas,r.;liu,d.l.;dewey,s.l.;silverman,r.b.,design and mechanism of(s)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid,a highly potent gamma-aminobutyric acid aminotransferase inactivator for the treatment of addiction.j.am.chem.soc.2018,140,2151-2164.
74.
[0321]
根据本公开内容,在结构上、在立体化学上和/或在构型上不同的各种其它化合物可通过这样的并入的合成程序和技术或其直接修改获得,这样的修改也将为本领域技术人员所知和理解并且意识到本发明,这样的程序、技术和修改仅受限于任何相应试剂或起始材料的商业或合成可用性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1