使用锇酸盐(VI)使烯烃二羟基化的制作方法
使用锇酸盐(vi)使烯烃二羟基化
1.相关申请的交叉引用
2.本技术要求于2019年11月4日提交的美国申请序列号62/930,280的优先权,将其内容通过援引以其整体并入。
技术领域
3.本发明涉及一种用于通过烯烃的二羟基化制备二醇化合物的方法,该方法提供了通过α-柏木烯的顺式二羟基化高效合成α,α-柏木烷二醇。
背景技术:
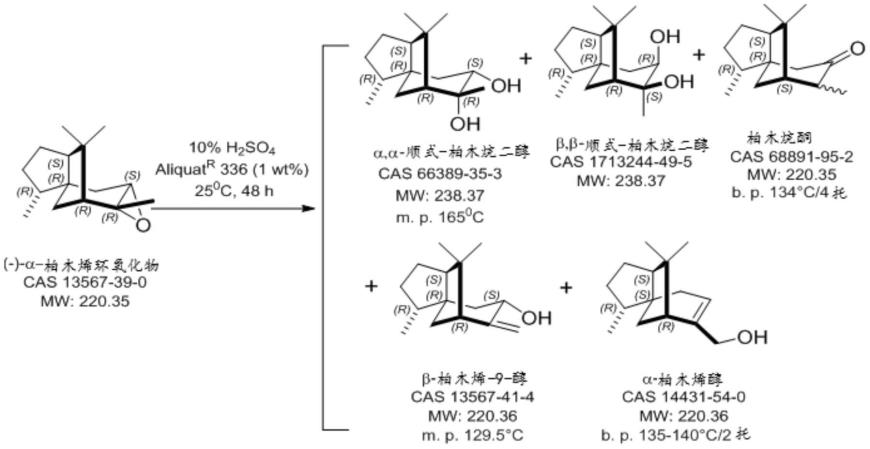
4.α,α-柏木烷二醇是合成(4ar,5r,7as,9r)-八氢-2,2,5,8,8,9a-六甲基-4h-4a,9-桥亚甲基甘菊环并(5,6-d)-1,3-间二氧杂环戊烯(极其强烈的木香-琥珀香韵)的中间体。已经报道了用于制备α,α-柏木烷二醇的两种方法。一种方法是基于α-柏木烯环氧化物的酸催化水解,并且另一种方法是α-柏木烯的直接四氧化锇催化二羟基化。α-柏木烯环氧化物的水解通常提供产物的混合物,并且α,α-柏木烷二醇的分离涉及繁琐的操作并且通常导致低产率。
5.例如,报道了(-)-α-柏木烯环氧化物在20℃下在4%的h2so4水溶液存在下的水解产生差向异构体的柏木烯酮(cedrenone)和二醇(推测为α,β-柏木烷二醇(recherches[研究],第16卷,第104-6页(1967)))。在10%的h2so4水溶液和aliquote r336存在下进行的水解(-)-α-柏木烯环氧化物的改进方法产生非对映异构体的混合物,产率仅为37.6%(美国专利号5892062)。美国专利号5892062公开了制备α-柏木烯环氧化物的两种方法,一种方法是通过在乙醚中在乙酸钠存在下用40%过乙酸进行环氧化,并且另一种方法是通过使用ad-混合物(ad-mix)的标准的锇催化不对称二羟基化(j.org.chem.[有机化学杂志],57(10),第2768-2771页(1992))。然而,本发明人按照所报道的方法进行的尝试并没有取得多大成功。根据美国专利号5892062的实例4中公开的反应条件的α-柏木烯环氧化物水解接着gc-ms分析表明所需的α,α-柏木烷二醇伴随有四种副产物:
[0006][0007]
这种组合物也描述于wo 2017186973中。将混合物从乙醇水溶液中重复重结晶产生α,α-柏木烷二醇,产率仅为5%-6%;因此,该方法不适用于所需二醇的经济制造。
[0008]
报道了通过化学计量的四氧化锇二羟基化α-柏木烯制备α,α-柏木烷二醇仅从屏蔽最少的一侧进行(j.org.chem.[有机化学杂志]ussr.8[6],1190(1972))。α-柏木烯与一当量的oso4在吡啶(py)中在20℃下反应4天产生熔点为159℃-160℃的α,α-柏木烷二醇,分离产率为31%。criegee(justus liebigs ann.chem.[利比希化学纪事],550[1],99(1942))报道了吡啶显著催化烯烃与四氧化锇的反应,包括庞大的烯烃二亚联苯基乙烯、四苯基乙烯和菲,并在环己烷-乙醇中用吡啶处理oso4分离出oso3·
2py络合物。
[0009]
upjohn公司(美国专利号2769824)首次报道了使用n-甲基吗啉氧化物(nmo)或其他脂肪族叔胺氧化物作为再氧化剂的孕二烯(pregnadien)的高效区域选择性四氧化锇催化的二羟基化。在叔丁醇和水中在1-5mol%oso4的存在下的反应需要在室温下几天才能实现部分转化。尽管发现添加水和吡啶对催化效率有一些有益的影响,但吡啶影响不能归因于吡啶n-氧化物的原位形成,因为后者是非反应性的再氧化剂。
[0010]
通过调整溶剂组成,upjohn报道了(tet.let[四面体快报],17[23]1973(1976))反应时间可以大大缩短:
[0011][0012]
这个过程被称为upjohn二羟基化。在类似条件下,其他简单烯烃也以合理至良好的产率转化为相应的顺式二醇化合物。
[0013]
在空间受阻诺甫醇衍生物的oso4催化二羟基化中,发现添加吡啶只有再氧化剂是三甲胺氧化物时才是有益的(tet.let.[四面体快报]21[5]449(1980))。在这些条件下,受阻α-蒎烯仅在β-侧二羟基化以在当量吡啶存在下在叔丁醇水溶液中回流4天后以96.5%的产率提供顺式-α-蒎烷二醇。相比之下,当nmo用作再氧化剂时,吡啶不仅没有提高反而大大降低了蒎烷二醇的相对平庸的产率(j.ind.chem.soc.[印度化学会志]59[2]119(1982))
[0014][0015]
(+)-蒎烷二醇是手性合成硼替佐米的关键中间体(千禧制药公司(millennium pharmaceuticals)的us 7714159),硼替佐米是经fda批准用于治疗多发性骨髓瘤的抗癌药物。
[0016]
使用三甲胺n-氧化物作为再氧化剂的α-柏木烯的oso4催化的顺式二羟基化首次报道于syn.commun.[合成通讯],28[20],3757(1998):
[0017][0018]
该反应需要3天以在沸腾的叔丁醇-水混合物中在过量吡啶存在下完成,尽管获得了不错的产率,产生具有m.p.162℃-163℃和[α]
d30-27.6
°
的产物(c 1.8,ch3oh)。
[0019]
此程序不适用于经济地制造α,α-柏木烷二醇,因为反应时间很长,四氧化锇是高毒性的,并且反应需要大量的吡啶,吡啶是必须回收利用的有毒化合物。此外,由于在分离产物时反应混合物必须用10倍体积的水稀释,因此产生大量有毒废物流,所以生产量低。
[0020]
在chemcatchem[催化化学],7[6]907(2015)中报道了使用商业50%的nmo水溶液作为再氧化剂的类似反应条件:
[0021][0022]
该反应也需要3天来完成,并且涉及在含有过量吡啶的沸腾叔丁醇水溶液中使用有毒的四氧化锇。
[0023]
因此,仍然需要开发一种更高效且更安全的工业方法来制造这种重要的香料产品。
技术实现要素:
[0024]
本发明通过提供一种经济的、可规模化的用于制备α,α-柏木烷二醇的方法来满足上述需要,并且本发明总体上适用于制备其他含有二醇官能团的精细化学品中的烯烃的二羟基化。
[0025]
在一个方面,本发明提供一种由烯烃制备二醇化合物的方法,该方法包括在催化
量的锇酸盐(vi)存在下在一种或多种溶剂中在高温下用氧化剂氧化该烯烃。
[0026]
在一些实施例中,有时优选地,锇酸盐(vi)是锇酸钾或锇酸钠二水合物,具有式m2oso4·
2h2o,其中m是k或na,其可以以m2oso2(oh)4的形式存在。
[0027]
在一些实施例中,该一种或多种溶剂选自由以下组成的组:c
1-c6脂肪族醇、其混合物、以及其与水的混合物。在一些优选的实施例中,脂肪族醇选自正丁醇、仲丁醇、异丁醇、叔丁醇、戊醇及其混合物。
[0028]
在一些实施例中,氧化剂是叔胺n-氧化物,包括但不限于三甲胺n-氧化物、三丁胺n-氧化物、n-甲基吗啉n-氧化物等。
[0029]
在一个具体方面,本发明提供了一种制备α,α-柏木烷二醇的方法,该方法包括在催化量的锇酸钾二水合物(k2oso4·
2h2o)的存在下在包含丁醇、优选异丁醇的溶剂中在回流温度下用n-甲基吗啉n-氧化物将α-柏木烯二羟基化。产物α,α-柏木烷二醇可以通过冷却从反应混合物中结晶并且过滤分离出来。
[0030]
在催化有效量的锇酸钾二水合物和作为再氧化剂的n-甲基吗啉-n-氧化物水溶液存在下在沸腾的异丁醇中的α-柏木烯的顺式二羟基化在仅24小时内就能以高产率和高立体选择性提供α,α-柏木烷二醇。
[0031]
使用异丁醇作为溶剂可以显著减少反应时间的发现是出人意料的并且不能从现有技术中推断出来。这似乎是第一次使用脂肪族伯醇作为二羟基化的溶剂。与上述文献方法中使用高挥发性和/或毒性试剂如oso4和吡啶相比,使用无毒且非挥发性锇酸钾二水合物允许安全地大规模制造α,α-柏木烷二醇。
[0032]
鉴于以下具体实施方式、实例和权利要求,本发明的其他方面和优点对于本领域普通技术人员将更加明显。
具体实施方式
[0033]
本发明基于以下出人意料的发现:锇酸盐(vi),例如锇酸钾(vi)二水合物(k2oso4·
2h2o),可用作烯烃二羟基化中oso4的安全替代物,其特别可用于通过α-柏木烯的二羟基化制备α,α-柏木烷二醇(合成(4ar,5r,7as,9r)-八氢-2,2,5,8,8,9a-六甲基-4h-4a,9-桥亚甲基甘菊环并(5,6-d)-1,3-间二氧杂环戊烯的前体)。
[0034]
在一个方面,本发明提供一种由烯烃制备二醇化合物的方法,该方法包括在催化量的锇酸盐(vi)存在下在一种或多种溶剂中在高温下用氧化剂氧化该烯烃。
[0035]
在一个实施例中,锇酸盐(vi)具有通式mjoso4,其中m是金属离子或铵离子,并且j是1或2,其可以以水合物mjoso4·
xh2o的形式存在,其中x选自1至6、优选为2(当j是1或2时),例如mjoso4·
2h2o或mjoso2(oh)4。
[0036]
在另一个实施例中,在式mjoso4、mjoso4·
2h2o、或mjoso2(oh)4的锇酸盐中,m是钾(k)或钠(na),并且j是2。
[0037]
在另一个实施例中,有时优选地,锇酸盐(vi)是k2oso4·
2h2o或k2oso2(oh)4。
[0038]
在一个实施例中,氧化剂是叔胺n-氧化物。
[0039]
在一个实施例中,叔胺n-氧化物具有通式r1r2r3n
+-o-,其中r1、r2和r3各自独立地是c
1-c
10
烷基、c
3-c8环烷基、或3-至10-元杂环基;或其中r1是c
1-c
10
烷基,并且r2和r3与它们所附接的n原子一起形成任选地含有1或2个选自o和n的额外的杂原子的5-至7-元杂环基。
[0040]
在另一个实施例中,有时优选地,氧化剂是n-甲基吗啉n-氧化物(nmo),有时更优选地nmo的水溶液。
[0041]
在另一个实施例中,溶剂包括脂肪族醇,并且有时优选地脂肪族醇和水的混合物。
[0042]
在另一个实施例中,脂肪族醇选自c
1-c8醇、其异构体、以及其混合物,有时优选地c
4-c5醇,并且有时更优选地异丁醇。
[0043]
在另一个实施例中,高温是在从35℃至一种或多种溶剂的回流温度的范围内。
[0044]
在另一个实施例中,溶剂是水和一种或多种c
1-c
10
脂肪族醇的混合物,并且高温是混合物的共沸温度(azeotrope temperature)。
[0045]
在另一个实施例中,醇选自由以下组成的组:甲醇、乙醇、正丙醇、异丙醇、正丁醇、异丁醇、仲丁醇、叔丁醇、戊醇、异戊醇及其混合物。
[0046]
在另一个实施例中,有时优选地,溶剂是异丁醇。
[0047]
在另一个实施例中,基于烯烃,催化量的锇酸盐(vi)是在约0.05mol%至约5mol%的范围内。
[0048]
在另一个实施例中,基于烯烃,催化量的锇酸盐(vi)是在约0.1mol%至约2mol%的范围内。
[0049]
在另一个实施例中,基于烯烃,催化量的锇酸盐(vi)是在约0.2至约1mol%的范围内。
[0050]
在另一个实施例中,有时优选地,基于烯烃,催化量的锇酸盐(vi)是在约0.3至约0.5mol%的范围内。
[0051]
在另一个实施例中,高温是在从约35℃至约150℃的范围内。
[0052]
在另一个实施例中,高温是在从约50℃至约125℃的范围内。
[0053]
在另一个实施例中,高温是在从约75℃至约115℃的范围内。
[0054]
在另一个实施例中,高温是水-醇共沸物的沸点。
[0055]
在另一个实施例中,有时优选地,溶剂是水和异丁醇的混合物,并且高温是93℃-95℃。
[0056]
在另一个实施例中,烯烃选自由以下组成的组:柏木烯、瓦伦烯、异胡薄荷醇、泪柏醇、香紫苏醇、α和β-蒎烯、莰烯、月桂烯、罗勒烯、d-柠檬烯、二戊烯、1-甲基环己烯、乙烯基环己烷、愈创木酚烯丙基醚、苯基烯丙基硫醚、乙烯基三甲基硅烷、3-(叔丁基二甲基甲硅烷基氧基)-2-甲茚、3-次乙基-1,5-二氢-2,4-苯并二氧杂环庚烷、茋、角鲨烯、(2e,4e)-己-2,4-二烯、6-甲基-1,5-庚二烯、2,6-二甲基庚-1,5-二烯、1,5,9-环十二碳三烯、异松油烯、α-松油醇、β-松油醇、δ-松油醇、烯丙醇、乙酸烯丙酯、烯丙基氯、β-香茅醇、羟基香茅醇、沉香醇、脱氢沉香醇、香叶醇、丁子香酚、β-水芹烯、α-侧柏烯、δ
3-蒈烯、橙花叔醇、反式-β-法尼烯、二氢法尼醇、法尼醇、高法尼醇β-石竹烯、α-红没药醇、2,4-癸二烯-1-醛、1-乙酰基环己烯、异弗洛瑞芬(isofloriffone)(1-[(1r,2s)-2,6,6-三甲基环己-3-烯-1-基]乙酮)、6-甲基-5-庚烯-2-酮、丙烯酸乙酯、山梨酸乙酯、(z)-己-3-烯-1-醇(叶醇)。
[0057]
在另一个实施例中,有时优选地,烯烃是柏木烯,并且二醇化合物是α,α-柏木烷二醇。
[0058]
在另一个实施例中,有时优选地,烯烃是柏木烯,并且异丁醇:柏木烯的重量比是在约0.5至约4的范围内,优选是约2。
[0059]
在另一个实施例中,该方法进一步包括反应的后处理和二醇化合物的分离。
[0060]
在另一个实施例中,分离包括结晶、分馏或色谱法。
[0061]
在一个具体方面,本发明提供了一种制备α,α-柏木烷二醇的方法,该方法包括在催化量的锇酸盐(vi)存在下在一种或多种溶剂中在高温下用氧化剂氧化柏木烯。
[0062]
在该具体方面的一个实施例中,锇酸盐(vi)是锇酸钾二水合物k2oso4·
2h2o。
[0063]
在该具体方面的一个实施例中,氧化剂是n-甲基吗啉n-氧化物。
[0064]
在该具体方面的一个实施例中,溶剂包括水和c
1-c6脂肪族醇,并且高温是在约75℃至115℃的范围内。
[0065]
在该具体方面的一个实施例中,溶剂是异丁醇,并且高温是93℃-95℃。
[0066]
在该具体方面的一个实施例中,该方法进一步包括通过以下方式分离α,α-柏木烷二醇:通过冷却反应混合物从反应混合物中结晶并过滤以收集结晶产物。
[0067]
为了说明,当在upjohn二羟基化条件下使用时,关于α-柏木烯,在叔丁醇和丙酮的混合物中使用0.3-0.4mol%k2oso4
·
2h2o和商业50%的nmo水溶液,在室温下观察到缓慢的反应,只产生10%的所需二醇,但选择性高。将温度提高到50℃,48小时后转化率提高到23%。
[0068]
使用dmf或n,n-二甲基乙酰胺作为溶剂并加热至85℃-90℃大大改进了转化率,在20小时后提供了85%的所需二醇(伴随着相应的不需要的酮醇)。
[0069]
当使用不同脂肪族醇作为反应溶剂时,观察到不同的结果。例如,使用沸腾的乙醇或异丙醇产生干净的反应,但在24小时后仅观察到部分转化。
[0070]
当叔丁醇用作唯一溶剂时,反应在85℃沸腾74小时后产生68%的转化率,并观察到约18%的副产物。
[0071]
出人意料地,当异丁醇用作溶剂时,加热至93℃-95℃持续24小时产生高选择性反应,转化率为96%-97%。事实上,仅形成了α,α-柏木烷二醇,如气相色谱法结果示出的,没有检测到β,β-柏木烷二醇的痕迹。也就是说,锇催化剂仅从受阻较小的底部侧(α攻击)接近α,α-柏木烯:
[0072][0073]
当使用沸腾的戊醇(混合异构体)时,获得了类似的结果。使用该溶剂混合物,反应在24小时后完成。反应混合物的沸点由具体的水-醇共沸物决定。这似乎是关于脂肪族伯醇作为锇催化烯烃二羟基化的反应溶剂的有益用途的第一份报道。
[0074]
添加吡啶(py)没有加速反应,并且当使用大量过量的吡啶时,反应变得更慢,推测
是由于催化剂转化为反应性较低的oso3·
2py。
[0075]
通过将反应混合物冷却至10℃持续1小时,以81%的产率获得所需的结晶α,α-柏木烷二醇。当温度降至2℃持续3小时时,过滤结晶产物并在室温下与己烷一起搅拌30分钟。以定量产率获得α,α-柏木烷二醇,其具有166.7℃的熔点。
[0076]
合适的溶剂是丁醇、异丁醇、仲丁醇、异丁醇和戊醇(异构体的混合物)。优选的溶剂是异丁醇。
[0077]
异丁醇:柏木烯的比率可以从0.5至4变化,优选的比率为2。
[0078]
k2oso4·
2h2o的负载量可以从0.1摩尔%至2摩尔%变化,更优选的范围是0.2摩尔%至0.6摩尔%,并且还优选的范围是0.38摩尔%。
[0079]
温度范围从75℃至115℃变化,优选范围是水-醇共沸物的沸点,即93℃-95℃。
[0080]
将新的反应条件应用于其他两种底物。当将具有85%纯度的瓦伦烯根据新的反应条件在沸腾的异丁醇中二羟基化时,24小时后以定量产率获得瓦伦烯-11,12-二醇。
[0081][0082]
据报道(wo 2006128126 a1)在丙酮水溶液中在室温下在1mol%oso4和作为再氧化剂的nmo存在下进行瓦伦烯的二羟基化。以未指定的产率完成转化。
[0083][0084]
当对异胡薄荷醇应用新的二羟基化时,仅回流12小时后以定量产率作为单一异构体获得对薄荷烷-3,8.9-三醇。根据helv.chim.acta[瑞士化学学报],87[10],2602(2004),对异胡薄荷醇应用夏普利斯(sharpless)ad-混合物二羟基化是不令人满意的,产生c8差向异构体的混合物。
[0085]
使用异丁醇/异戊醇的回流混合物将新的二羟基化条件应用于(+)-α-蒎烯。反应在14小时内完成,以单一异构体的形式定量地提供(+)-蒎烷二醇。
[0086][0087]
本技术中的任何术语,除非特别定义,否则均采用本领域普通技术人员所理解的普通含义。
[0088]
如本文所用的,单数形式“一种/个(a/an)”和“该(the)”包括复数个指示物,除非上下文中另外明确指明。
[0089]
如本文所用的,术语“约”通常包括指示数字的最高达正或负10%。例如,“约10%”可能指示9%至11%的范围,并且“约20”可能意指从18至22。有时优选地,术语“约”包括指示值的最高达正或负5%。
[0090]
除非另有说明,否则在此和权利要求书中提及的所有份数、百分比和比例均以重量计。
[0091]
如本文所公开的,提供了多个数值范围。应理解,该范围的上限与下限之间的每个中间值(至下限的个位的十分之一,除非上下文另外明确指明)也被具体披露。
[0092]
短语“反应器”是指实际进行反应的装置。
[0093]“烷基”是指饱和脂肪族烃基,包括c
1-c
12
直链和支链基团。优选烷基是具有1至8个、有时更优选1至6个碳原子的烷基。代表性实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1-二甲基丙基、1,2-二甲基丙基、2,2-二甲基丙基、1-乙基丙基、2-甲基丁基、3-甲基丁基、正己基、1-乙基-2-甲基丙基、1,1,2-三甲基丙基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2-乙基丁基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2,3-二甲基丁基等。
[0094]“环烃基”是指具有3至8个碳原子、有时优选3至6个碳原子的饱和和/或部分不饱和的单环烃基。单环环烃基的代表性实例包括但不限于环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环己二烯基、环庚基、环庚三烯基、环辛基等。
[0095]“杂环基”是指具有一个或多个选自由n和o组成的组的杂原子的3至10-元、有时优选5-至6-元、饱和和/或部分不饱和的单环或多环烃基。单环杂环基的代表性实例包括但不限于吡咯烷基、哌啶基、哌嗪基、吗啉基等。
[0096]
以下非限制性实例用于说明本发明的某些方面。
[0097]
实例
[0098]
实例1.
[0099]
向配备有机械搅拌器和回流冷凝器的1l反应器中添加工业级(-)-α-柏木烯(110g,91%纯度,0.49mol,[α]
d20
=-84.2
°
,纯的)、异丁醇(200g)、锇酸钾二水合物(0.7g,1.9mmol,0.39mol%)和在水中50wt%的n-甲基吗啉n-氧化物(235g,1mol)。将反应混合物加热至93℃-95℃,在该温度下保持温和回流24小时。gc分析表明仅剩下3%-4%未反应的α-柏木烯,并且仅获得了顺式柏木烷二醇的单一非对映异构体。
[0100]
将反应混合物在2℃下冷却3小时以获得结晶产物,将其过滤并用水洗涤。将晶体
转移到反应器中,向其中添加3份己烷,并将混合物在室温下搅拌30分钟以提供白色晶体。因此,通过过滤和干燥,以几乎定量的产率和99.5%的纯度获得115g所需产物,其具有m.p.166.1℃(lit.165℃)和[α]
d20
=-21.180(c 0.7%chcl3)。nmr光谱与文献数据(chemcatchem[催化化学],7[6]907(2015)支持信息)相符。
[0101]
实例2.
[0102]
向配备有机械搅拌器和回流冷凝器的1l反应器中添加(-)-α-柏木烯(110g,91%纯度,0.49mol,[α]
d20
=-84.20,纯的)、异丁醇(200g)、锇酸钾二水合物(1g,2.68mmol,0.54mol%)和在水中50wt%的n-甲基吗啉n-氧化物(200g,0.853mol)。将反应混合物加热至93℃-95℃,在该温度下保持温和回流24小时。gc分析表明仅剩下3%-4%未反应的α-柏木烯,并且仅获得了顺式柏木烷二醇的单一非对映异构体。
[0103]
在1小时期间将反应混合物冷却至10℃以获得结晶产物,将其在布氏漏斗上过滤并用水洗涤。将晶体转移到反应器中,向其中添加3份己烷,并将混合物在室温下搅拌30分钟以提供白色晶体。因此,通过过滤和干燥以81%产率和99.5%纯度获得了95g所需产物,其具有m.p.166.1℃(lit.165℃)。
[0104]
实例3.
[0105]
向配备有机械搅拌器和回流冷凝器的1l反应器中添加α-柏木烯(110g,91%纯度,0.49mol,[α]
d20
=-84.20,纯的)、异戊醇(100g,异构体的混合物,默克(merck)b.p.131℃)、锇酸钾二水合物(0.7g,0.39mol%)和在水中50wt%的nmo(235g,1.0mol)。将反应混合物加热至93℃-95℃,在该温度下保持温和回流24小时。gc分析表明仅剩下3%-4%未反应的α-柏木烯,并且仅获得了顺式柏木烷二醇的单一非对映异构体。
[0106]
在1小时期间将反应混合物冷却至10℃并过滤以获得结晶产物,将其收集在布氏漏斗上并用水洗涤。将晶体转移到反应器中,向其中添加3份己烷,并将混合物在室温下搅拌30分钟。以81%产率(95g)和99.5%纯度获得白色结晶产物,m.p.166.1℃(lit.165℃)。
[0107]
实例4.
[0108]
向配备有机械搅拌器和回流冷凝器的1l反应器中添加瓦伦烯(117.8g,85%纯度,0.49mol)、异丁醇(100g)、锇酸钾二水合物(0.7g,0.39mol%)和在水中50wt%的nmo(235g,1.0mol)。将混合物加热至93℃-95℃,在该温度下保持温和回流24小时。gc分析表明瓦伦烯完全转化为瓦伦烯-11,12-二醇。将反应混合物通过连二亚硫酸钠水溶液和处理,接着过滤。用12n h2so4将滤液酸化至ph 2,以将n-甲基吗啉转化为其硫酸氢盐。分离相,将水相用异丁醇萃取,并且将合并的有机相用25%nacl水溶液洗涤。减压蒸馏异丁醇。将残余物在真空下分馏以产生呈淡黄色油状物的所需产物。产率为95%。
[0109]
实例5.
[0110]
向配备有机械搅拌器和回流冷凝器的1l反应器中添加(-)-异胡薄荷醇(76g,99.5%纯度,0.49mol)、异丁醇(100g)、锇酸钾二水合物(0.7g,0.39mol%)和在水中50wt%的nmo(235g,1.0mol)。将反应混合物加热至93℃-95℃,在该温度下保持温和回流12小时。gc分析表明(-)-异胡薄荷醇完全转化为对薄荷烷-3,8.9-三醇的单一非对映异构体。反应混合物的常规后处理产生呈油状物的所需产物。分馏产生纯的对薄荷烷-3,8.9-三醇,在140℃-145℃/0.08托下沸腾。产率为93%。
[0111]
实例6.
[0112]
向配备有机械搅拌器和回流冷凝器的1l反应器中添加(+)-α-蒎烯(100g,92%纯度,0.67mol)、异丁醇(25g)、异戊醇(75g,异构体混合物,b.p.131℃)、锇酸钾二水合物(0.20g,0.08mol%)和在水中50wt%的nmo(175g,0.744mol)。将反应混合物加热至92℃,在该温度下保持温和回流14小时。gc分析表明(+)-α-蒎烯完全转化为(+)-蒎烷二醇的单一非对映异构体。将反应混合物用250ml水稀释并用己烷(2
×
200ml)萃取。将合并的有机相用100ml水洗涤。蒸发溶剂以产生白色固体。根据内标的产率为96%。
[0113]
出于所有目的,本文引用的所有出版物均通过援引以其整体并入。尽管在上面的实例中已经描述了几个实施例,但本领域普通技术人员将理解,在不背离如由以下权利要求限定的本公开的精神和范围的情况下,可以对其中的形式和细节进行各种改变。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1