一种清热解毒的绿原酸衍生物及其制备方法与流程
本发明属于化学药物合成制备
技术领域:
,涉及一种清热解毒的绿原酸衍生物及其制备方法。
背景技术:
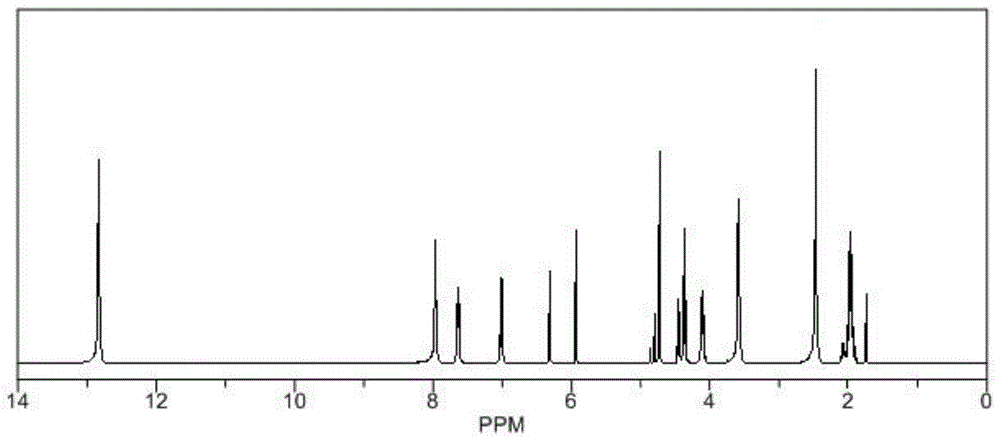
:绿原酸是植物体内在有氧呼吸过程中经莽草酸途径产生的一种苯丙素类化合物,自20世纪50年代首次从苹果中提取成功就一直受到学者们的高度关注。研究表明绿原酸具有清热解毒的功效,常被用在清热解毒的中药类制剂中。但是绿原酸分子中存在苯酚结构,苯酚常置于空气中容易发生氧化,使绿原酸的结构发生改变,变得不稳定,影响其生物活性,因此需要对绿原酸结构进行修饰,获得结构更加稳定的绿原酸衍生物,同时还能拥有更好的生物活性。技术实现要素:为了解决上述技术问题,本发明提供了一种清热解毒的绿原酸衍生物,其衍生物具有式(i)分子结构:本发明的另一个目的在于提供清热解毒的绿原酸衍生物的制备方法,包括如下步骤:在氮气保护下,室温下将绿原酸溶解于无水有机溶剂中,加入无水碱,再加入三甲基氯硅烷,室温下搅拌反应2~3h,tlc检测反应结束;在反应液中加入3,5-二甲基苯甲酰氯,搅拌溶解,升高温度至80~110℃,tlc检测反应结束,加入盐酸溶液,搅拌3~5h,用饱和食盐水洗涤,有机层用无水硫酸钠干燥,减压蒸馏除去溶剂,残留物硅胶柱洗脱层析得最终产物;所述绿原酸与三甲基氯硅烷的摩尔比为1:(1.0~4.5);所述绿原酸与3,5-二甲基苯酰氯的摩尔比为1:(1.5~3.5);所述碱为三乙胺、乙二胺、氢氧化钠、碳酸钠、醋酸钠或碳酸钾;所述有机溶剂为二氯甲烷、三氯甲烷、四氢呋喃、丙酮、乙腈或乙醚、苯或甲苯;所述硅胶柱洗脱液为石油醚与二氯甲烷的混合液,其中石油醚与二氯甲烷的体积比为(2~7):1。根据上述制备方法的一种优选,包括如下步骤:在氮气保护下,室温下将15g绿原酸溶解于无水120ml二氯甲烷中,加入无水三乙胺,再加入13.8g三甲基氯硅烷,室温下搅拌反应3h,tlc检测反应结束;在反应液中加入17.8g3,5-二甲基苯甲酰氯,搅拌溶解,升高温度至110℃,tlc检测反应结束,加入盐酸溶液,搅拌3h,用饱和食盐水洗涤,有机层用无水硫酸钠干燥,减压蒸馏除去溶剂,残留物以体积比为2:1的石油醚与二氯甲烷的混合液为洗脱液,将残留物经硅胶柱洗脱层析得最终产物。现有技术相比,本发明的有益效果是:本发明采用天然植物提取物绿原酸,对其结构改性,获得一种具有酰胺键的绿原酸衍生物;病理实验表明,绿原酸衍生物对大肠杆菌、表皮葡萄球菌表现出较好的抑制活性,尤其对大肠杆菌抑制活性更加显著,比单独使用的绿原酸的抑制效果更好;对h1n1流感病毒业具有显著的抑制作用;对抑制巴豆油致小鼠耳肿胀,其抑制效果与清开灵注射液相当;以及对伤寒、副伤寒甲乙三联菌苗所致家兔发热具有降低作用。总之,本发明的绿原酸衍生物具有发展为清热解毒药物的潜力。附图说明图1:实施例1的清热解毒的绿原酸衍生物核磁共振氢谱图。具体实施方式为了使本发明的目的、技术方案及优点更加清楚明白,下面结合具体实施方式对本发明作进一步的详细描述,但不应将此理解为本发明上述主题的范围仅限于下述实施例。实施例1在氮气保护下,室温下将15g绿原酸溶解于无水120ml二氯甲烷中,加入无水三乙胺,再加入13.8g三甲基氯硅烷,室温下搅拌反应3h,tlc检测反应结束;在反应液中加入17.8g3,5-二甲基苯甲酰氯,搅拌溶解,升高温度至110℃,tlc检测反应结束,加入盐酸溶液,搅拌3h,用饱和食盐水洗涤,有机层用无水硫酸钠干燥,减压蒸馏除去溶剂,残留物以体积比为2:1的石油醚与二氯甲烷的混合液为洗脱液,将残留物经硅胶柱洗脱层析得最终产物。产率为80.33%。核磁共振氢谱检测:将样品放入样品管中,用注射器取0.5mlcdcl3(氘代氯仿)注入样品管,使样品充分溶解。要求样品与试剂充分混合,溶液澄清、透明、无悬浮物或其他杂质,经核磁共振鉴定,得到核磁共振氢谱图,结果见如图1。实施例2本发明化合物对清热解毒的药效学试验1.本发明药物的体外抗菌活性实验供试菌种:金黄色葡萄球菌(a)、大肠杆菌(b)、枯草芽孢杆菌(c)、表皮葡萄球菌(d)、白色念珠球菌(e)。将本发明的绿原酸衍生物溶解在无水乙醇中,用胰蛋白胨大豆肉汤培养液稀释至1000μg/ml,继续用培养液稀释使药物的浓度从256~0.25μg/ml,获得实验溶液;再配制浓度范围为256~0.25μg/ml的清开灵注射液作为阳性对照组(对照组1);同样的方法配制绿原酸作为对照组2。在96孔板上每孔加入浓度范围为256~0.25μg/ml的药液和100μl的菌液,最终药物的浓度为5×104cfu,以胰蛋白胨大豆肉汤培养基加菌液作为阴性对照(tsb培养基、菌液各100μl),以不加菌液的tsb肉汤培养基为空白对照(tsb培养基200μl),将96孔板密封后置于37℃恒温培箱中,孵育20小时。测定各孔625nm处的吸光度光度值,与空白对照组od625值一致的孔视为细菌无明显生长。细菌无明显生长的药物最低浓度为本发明化合物的最小抑菌浓度mic。表1本发明化合物的体外抗菌活性测试由表1数据表明,绿原酸衍生物对大肠杆菌、表皮葡萄球菌表现出较好的抑制活性,尤其对大肠杆菌抑制活性更加显著,比单独使用的绿原酸的抑制效果更好。2.本发明化合物对感染h1n1流感病毒小鼠肺指数影响选取昆明小鼠若干,随机分组,分别为正常对照组(对照组1)、模型对照组(对照组2)、清开灵注射液(对照组3)、绿原酸组(对照组4)、实验组。小鼠每天灌喂给药1次,各组动物给药2天后,小鼠用乙醚轻度麻醉,以15个ld50病毒液滴鼻感染,每只小鼠0.5ml,小鼠苏醒后继续给药4天。正常对照组和病毒对照组给与等体积生理盐水。第5天称量体重,腋下动脉放血处死后取出鼠肺,计算肺指数和抑制率,肺指数值越大,表示肺炎越严重,结果见表2。肺指数抑制率=(病毒对照组肺指数均值-实验组肺指数均值)/病毒对照组肺指数均值×100%。肺指数=肺湿重(g)/体重表2本发明化合物对感染h1n1流感病毒小鼠肺指数影响组别剂量(g/kg)肺指数值抑制率(%)对照组1-0.79±0.07-对照组2-2.47±0.21-对照组30.091.12±0.1546.2对照组430.01.32±0.1832.1实验组30.01.20±0.2140.3由表2数据表明,本发明化合物能有效降低肺指数,肺指数抑制率为40.3%,高于单独作用的绿原酸抑制率(32.1%),由此说明,本发明的绿原酸衍生物对h1n1流感病毒具有显著的抑制作用。3.本发明化合物的抗炎作用实验选取健康的雄性昆明小鼠,随机分组为:模型组(对照组1)、清开灵注射液组(对照组2)、绿原酸组(对照组3)、实验组。每组动物每天给药一次,模型组灌喂等体积的蒸馏水,其他各组灌喂相对应的药物,连续灌喂三天,于末次给药1h后,在每只动物左耳涂抹巴豆油0.02ml,右耳不涂作为正常耳,致炎4h后处死,剪下两只耳朵,用直径0.8cm打孔器在耳同一部位打下耳片,分别称取两片耳重量,以重量差值作为肿胀程度。实验结果见表3。肿胀抑制率%=(模型组左右耳片重量差-给药组左右耳片重量差)/模型组左右耳片重量差×100%表3本发明化合物对巴豆油致小鼠耳肿胀的影响组别剂量(g/kg)肿胀度肿胀抑制率(%)对照组1-35.39±2.72-对照组215.023.16±3.0450.59对照组330.030.48±2.3727.51实验组30.025.61±2.2645.77由表3数据表明,与模型组相比,本发明的绿原酸衍生物能显著抑制巴豆油致小鼠耳肿胀,其抑制效果与清开灵注射液相当,且优于单独使用时的绿原酸的抑制效果。4.本发明化合物的解热作用实验测试本发明药物对伤寒、副伤寒甲乙三联菌苗所致家兔发热模型的影响。选取健康的家兔,随机分组。连续测3次肛温,每次3min,间隔1h,将3次结果的平均值作为基础体温,然后在兔子耳朵静脉注射伤寒、副伤寒甲乙三联菌苗1ml/kg,待肛温升高0.5℃时灌喂给药。实验组灌喂给药绿原酸衍生物15g/kg,模型组(对照组1)灌喂给予等量的蒸馏水,阳性对照组(对照组2)清开灵注射液,绿原酸组(对照组3)灌喂给予30g/kg绿原酸,每隔1h测温一次,连续6h。计算各组动物在给药后不同时间内的体温变化,实验结果见表4。表4本发明化合物的解热作用影响由表4数据表明,与模型对照组相比,三种药物均能降低感染伤寒、副伤寒甲、乙三联菌苗后家兔的体温,其中绿原酸衍生物与清开灵注射液的效果相当,且都远优于单独作用的绿原酸降温效果。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1