一种水稻尖线虫ATP合成酶及其应用
一种水稻尖线虫atp合成酶及其应用
技术领域
1.本发明涉及植物病虫害防治技术领域,具体地,涉及一种水稻尖线虫atp合成酶及其 应用。
背景技术:
2.水稻是最重要的粮食作物之一,植物寄生线虫每年可造成全球水稻逾160亿美元的经 济损失,其中最主要的寄生为害水稻地上部的线虫是水稻干尖线虫(aphelenchoidesbesseyi)。水稻干尖线虫侵染水稻,造成经济损失可高达50%。该线虫在我国主要稻区均 有分布且局部为害严重。
3.atpase基因与植物抗病诱发的过敏性坏死有重要关系。目前发现,稻瘟病菌 (magnaporthe grisea)的atpase蛋白基因pde1与稻瘟菌侵染时附着胞形成密切相关; 稻瘟病菌的另一个atpase基因mgapt2可以诱发植物产生抗性,而发生过敏性坏死。植 物也可以通过自身分泌调节atpase来激发自身防御反应。烟草atpase ntaaa1参与自身 抗病反应,沉默该基因后,接种丁香假单胞细菌(pseudomonas syringae)的烟草丧失了 过敏性坏死的防御反应。拟南芥的位于线粒体外膜的atpase atom66在诱导拟南芥发生 过敏性坏死抵御病原物侵染中起重要作用。据报道,atpase广泛存在于线虫体内,在线 虫的生命活动起着相当重要的作用。
4.在植物线虫中,目前只有南方根结线虫(meloidogyne incognita)的atpase基因被成 功克隆,据报道,沉默南方根结线虫的atp合成酶基因可导致线虫二龄幼虫死亡率显著 升高,体外浸泡和植物介导的rnai结果均表明沉默该基因导致致病性减弱,寄主的根结 数量显著降低(huang y h,mei m,shen b m,et al.rnai of miasb caused high mortality ofmeloidogyne incognita juveniles and inhibited the nematode diease[j].agricultural sciences, 2013,9(4):483
‑
490;huang y h,mei m,mao z c,et al.molecular cloning and virus
‑
inducedgene silencing of miasb in the southern root
‑
knot nematode,meloidogyne incognita[j]. european journal of plant pathology,2014,138:181
‑
193)。然而至今尚无水稻干尖线虫atp 合成酶及其功能的相关报道。
技术实现要素:
[0005]
本发明要解决的技术问题是克服现有水稻干尖线虫防治技术不足以及水稻干尖线虫 效应蛋白atp合成酶的研究不足,提供一种水稻尖线虫atp合成酶及其应用,本发明为 抗线虫品种植物的培育提供了新靶标,为水稻干尖线虫的防治提供了新途径。
[0006]
本发明的第一个目的是提供一种水稻尖线虫atp合成酶基因。
[0007]
本发明的第二个目的是提供一种水稻尖线虫atp合成酶。
[0008]
本发明的第三个目的是提供所述的水稻尖线虫atp合成酶基因的抑制剂和/或所述的 水稻尖线虫atp合成酶的抑制剂在制备防治水稻尖线虫药物中的应用。
[0009]
本发明的第四个目的是提供所述的水稻尖线虫atp合成酶基因的和/或所述的水稻尖 线虫atp合成酶作为抗水稻尖线虫药物筛选靶点的应用。
[0010]
本发明的第五个目的是提供一种所述的水稻尖线虫atp合成酶基因和/或所述的水稻 尖线虫atp合成酶的抑制剂。
[0011]
本发明的第六个目的是提供编码中所述的dsrna的基因。
[0012]
本发明的第七个目的是提供含有所述的基因的表达载体、转基因细胞系或宿主菌。
[0013]
本发明的第八个目的是提供所述的基因、和所述的表达载体、转基因细胞系或宿主菌 中的任意一种或几种在制备水稻尖线虫防治药品中的应用。
[0014]
为了实现上述目的,本发明是通过以下方案予以实现的:
[0015]
本发明要求保护一种水稻尖线虫atp合成酶基因,seq id no:1所示序列为水稻尖 线虫atp合成酶基因全长。
[0016]
以及,一种水稻尖线虫atp合成酶,其氨基酸序列如seq id no:2所示。
[0017]
所述的水稻尖线虫atp合成酶基因的抑制剂和/或所述的水稻尖线虫atp合成酶的抑 制剂在制备防治水稻尖线虫药物中的应用,也属于本发明的保护范围。
[0018]
优选地,所述药物抑制水稻干尖线虫的繁殖能力。
[0019]
优选地,所述药物抑制水稻干尖线虫的生长能力。
[0020]
优选地,所述抑制剂为dsrna。
[0021]
更优选地,所述dsrna为由如seq id no.3所示的核苷酸序列作为正义链及由与 seq id no.3所示的核苷酸序列反向互补的核苷酸序列作为反义链组成的双链rna。
[0022]
所述的水稻尖线虫atp合成酶基因的和/或所述的水稻尖线虫atp合成酶作为抗水稻 尖线虫药物筛选靶点的应用,也属于本发明的保护范围。
[0023]
本发明还要求保护一种所述的水稻尖线虫atp合成酶基因和/或所述的水稻尖线虫 atp合成酶的抑制剂,所述抑制剂为dsrna,所述dsrna为由如seq id no.3所示的 核苷酸序列作为正义链及由与seq id no.3所示的核苷酸序列反向互补的核苷酸序列作 为反义链组成的双链rna。
[0024]
本发明还要求保护,编码中所述的dsrna的基因;
[0025]
以及,含有所述的基因的表达载体、转基因细胞系或宿主菌。
[0026]
所述的基因、和所述的表达载体、转基因细胞系或宿主菌中的任意一种或几种在制备 水稻尖线虫防治药品中的应用,也属于本发明的保护范围。
[0027]
与现有技术相比,本发明具有以下有益效果:
[0028]
本发明提供了一种水稻尖线虫atp合成酶及其应用,采用rnai处理后,能够抑制水 稻尖线虫的繁殖力,促进水稻防卫基因表达并可激活植物免疫反应,可以诱导细胞凋亡, 为农业生产中的线虫防治提供一条新的策略。本发明为抗线虫品种植物的培育提供了新靶 标,为水稻干尖线虫的防治提供了新途径。
附图说明
[0029]
图1为水稻干尖线虫和其他线虫atp合成酶的系统进化树;下划线处为水稻干尖线 虫ab
‑
atps;每个序列的基因号在括号内标注。
[0030]
图2为ab
‑
atps在水稻干尖线虫卵、幼虫、雌虫和雄虫中的表达量;
“ⅰ”
表示平均 数的标准误(n=3),不同字母代表各处理间差异显著(p<0.05)。
[0031]
图3为水稻干尖线虫ab
‑
atps基因编码的mrna的组织定位;a:ab
‑
atps s mrna定 位于食道;c:ab
‑
atps s mrna定位于生殖系统;b、d:正义探针对照;oesophagus:食 道;mb:中食道球;v:阴门;reproductive system:生殖系统。
[0032]
图4为不同处理后水稻干尖线虫体内ab
‑
atps基因的沉默效率检测;g12、g24、g36 和g48:gfp dsrna处理线虫12h、24h、36h和48h;a12、a24、a36和a48:ab
‑
atps dsrna 处理线虫12h、24h、36h和48h;
“ⅰ”
表示平均数的标准误(n=3),不同字母代表各处 理间差异显著(p<0.05)。
[0033]
图5为水稻osrlk3和水稻干尖线虫ab
‑
atps的互作;a:osrlk3和ab
‑
atps的互 作;pgbkt7
‑
osrlk3(bd)与pgadt7
‑
ab
‑
atps(ad)共转化的酵母在ydpa上生 长为白色菌落,在qdo/xa(sd/
‑
leu/
‑
trp/
‑
ade/
‑
his/x
‑
α
‑
gal/aba)培养基上生长,培养 基变蓝;共转pgadt7
‑
lam和pgbkt7
‑
53酵母作为阳性对照;共转pgadt7空载体和 pgbkt7
‑
osrlk3酵母,pgadt7
‑
ab
‑
atps和pgbkt7空载体酵母作为阴性对照;b: pgbkt7
‑
osrlk3(bd)与pgadt7
‑
ab
‑
atps(ad)共转化的酵母菌液经梯度稀释1, 10
‑1,10
‑2在qdo/xa培养基上生长。
[0034]
图6为不同处理后的水稻干尖线虫繁殖量和osrlk3基因表达变化;a:g12,g24,g36 和g48:gfp dsrna处理线虫12h、24h、36h和48h后接种培养基35d后分离;a12、 a24、a36和a48:ab
‑
atps基因rnai处理水稻干尖线虫12h、24h、36h和48h后接种 培养基35d后分离到的线虫数,
“ⅰ”
表示平均数的标准误(n=9),不同字母代表各处 理间差异显著(p<0.05);b:柱高为处理样品和空白对照的比值的平均数(n=3,每个重 复包括三株水稻,技术重复二次)。内参基因为osubq5,
“ⅰ”
表示平均数的标准误(n=3), 不同大写字母代表接种ab
‑
atps dsrna浸泡48h的水稻干尖线虫水稻的osrlk3基因在各 处理间差异显著(p<0.05),不同小写字母代表接种gfp dsrna浸泡48h的水稻干尖线虫 水稻的osrlk3基因在各处理间差异显著(p<0.05),*:同一时间接种ab
‑
atps dsrna 浸泡48h的水稻干尖线虫水稻与接种gfp dsrna浸泡48h的水稻干尖线虫水稻的基因表达 水平存在显著差异(p<0.05),ns:同一时间接种ab
‑
atps dsrna浸泡48h的水稻干尖线 虫水稻与接种gfp dsrna浸泡48h的水稻干尖线虫水稻的基因表达水平无显著差异 (p>0.05),dai:接种后天数,gfp:接种gfp dsrna浸泡48h的水稻干尖线虫水稻,ab
‑
atps: 接种ab
‑
atps dsrna浸泡48h的水稻干尖线虫水稻。
[0035]
图7为水稻干尖线虫ab
‑
atps在烟草中的亚细胞定位和瞬时表达;a:ab
‑
atps在 烟草中定位于细胞膜、细胞质和细胞核;b:western
‑
blot检测ab
‑
atps在烟草中表达, m:低分子量蛋白marker,1:gfp在烟草中的瞬时表达,2:ab
‑
atps
‑
gfp在烟草中的 瞬时表达;c:ab
‑
atps引起烟草叶片细胞死亡,1:mes缓冲液,2:pcambia1300
‑
gfp, 3:pcambia1300
‑
ab
‑
atps
‑
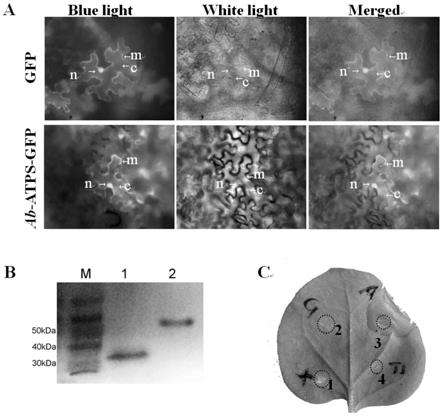
gfp,4:pcambia1300空载体质粒。
具体实施方式
[0036]
下面结合说明书附图及具体实施例对本发明作出进一步地详细阐述,所述实施例只用 于解释本发明,并非用于限定本发明的范围。下述实施例中所使用的试验方法如无特殊说 明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂 和材料。
[0037]
实施例1水稻尖线虫atp合成酶ab
‑
atps基因全长序列的克隆和序列分析
[0038]
一、实验方法
[0039]
利用水稻的一个关键防卫基因osrlk3与水稻干尖线虫的cdna文库通过酵母双杂交 实验钓取了基因,经过测序得到了水稻干尖线虫ab
‑
atps基因的部分序列。
[0040]
进一步通过这一序列设计race引物,然后以水稻干尖线虫cdna为模板,使用bd smarttm pcr cdna synthesis kit(takara,japan),来获得水稻干尖线虫ab
‑
atps的基 因全长,具体步骤如下:
[0041]
1、5
′
race扩增:
[0042]
(1)第一轮pcr引物:
[0043]
正向引物upm:5
’‑
ctaatacgactcactcactatagggc
‑3’
[0044]
反向引物d2racer1:5
’‑
atcgccaatctgaacgatcaagccgcccat
‑3’
[0045]
反应体系:
[0046][0047]
反应条件:94℃反应30s,72℃反应3min,5个循环;94℃反应30s,70℃反应30s, 72℃反应3min,5个循环;94℃反应30s,68℃反应30s,72℃反应3min,25个循环; 72℃反应10min;4℃保存。
[0048]
反应结束后用1%琼脂糖凝胶电泳检测,剩余pcr产物置于
‑
20℃保存。
[0049]
(2)第二轮巢式(nest)pcr引物:
[0050]
正向引物nup:5
’‑
aagcagtggtatcaacgcagagt
‑3’
[0051]
反向引物ia
‑
d2r:5
’‑
ccgactgtttcaagttgcgat
‑3’
[0052]
反应体系:
[0053][0054]
反应条件:94℃反应30s,72℃反应3min,5个循环;94℃反应30s,70℃反应30s, 72
℃反应3min,5个循环;94℃反应30s,68℃反应30s,72℃反应3min,25个循环; 72℃反应10min;4℃保存。
[0055]
反应结束后用1%琼脂糖凝胶电泳检测,剩余pcr产物置于
‑
20℃保存。
[0056]
2、pcr产物连接转化
[0057]5’
race扩增产物纯化后用pmd 18
‑
t载体(takara,japan)连接以获得重组质粒,并 转化到大肠杆菌(escherichia coli)感受态细胞dh5α中,挑取阳性克隆送到生工生物公 司测序(sangon biotech,china)。
[0058]
3、ab
‑
atps基因全长序列扩增
[0059]
根据测序结果获得的目的基因的3’poly a序列,设计引物用于水稻干尖线虫ab
‑
atps 基因cdna进行全长扩增。
[0060]
pcr引物序列:
[0061]
d2f:5
’‑
tttgagcgaaatgtccgcaag
‑3’
[0062]
d2r:5
’‑
agtctacgagttcctactgacga
‑3’
[0063]
反应体系:
[0064][0065]
反应条件:94℃反应2min;98℃反应10s,55℃反应30s,72℃反应15s,30个循环; 72℃反应10min。
[0066]
用mega(molecular evolutionary genetics analysis,usa)软件的neighbor
‑
joining 法构建ab
‑
atps和数据库中其他23个代表性寄生虫的atp合成酶氨基酸序列的系统进化 树。
[0067]
二、实验结果
[0068]
提取阳性克隆质粒并测序,得到301bp的含polya片段。将该基因进行race扩增, 获得全长为1341bp的cdna序列(如seq id no:1所示),用ncbi的orf finder进 行orf识别,得到完整的orf,该基因编码183个氨基酸(如seq id no:2所示)。
[0069]
将水稻干尖线虫ab
‑
atps和蛋白数据库中23个代表性线虫的atp合成酶氨基酸序列 进行比对并构建系统进化树(图1),结果显示,水稻干尖线虫ab
‑
atps与拉弟类圆线虫 (strongyloides ratti)的atp合成酶亲源关系最近,这与blastx得到的同源性分析结果 一致。
[0070]
实施例2 ab
‑
atps基因在水稻干尖线虫不同虫态的表达量测定
[0071]
一、实验方法
[0072]
1、使用microelute total rna kit(omega)分别提取供试水稻干尖线虫的500条雌 虫、雄虫、幼虫和卵的rna。具体步骤如下:
[0073]
使用microelute total rna kit试剂盒,并按照说明书操作进行微量线虫rna提取, 步骤如下:
[0074]
(1)根据需要将线虫(<500条)放入depc处理过的1.5ml离心管中,用depc 水清洗3次。将离心管置于预冷的研钵中,往研钵中加入液氮并立即用研磨棒将管中的线 虫研磨成细粉状,快速加入350μl试剂trk、7μlβ
‑
巯基乙醇。
[0075]
(2)涡旋振荡器振荡30s,1000rpm离心1min,吸取上清液到另一个新的depc处 理的1.5ml离心管中。
[0076]
(3)向装有上清液的新管中加入350μl 70%乙醇,均匀混合。
[0077]
(4)将混合液加入试剂盒提供的硅胶过滤柱套管,1000rpm低速离心1min。
[0078]
(5)倒弃滤液,将过滤柱移至新的2ml离心管上,加入400μl rwc溶液,10000rpm 离心1min。
[0079]
(6)均匀混合30μl dnase
‑
i、7μl buffer和33μl depc水,加到过滤柱的滤膜中心, 37℃30min。根据是否需要去除rna中的dna,决定是否完成此步骤。
[0080]
(7)将过滤柱移至新的2ml离心管上,加入500μl溶液,10000rpm离心1min。
[0081]
(8)重复上一步。
[0082]
(9)全速离心(13000rpm)过滤柱空管,使柱子上的滤膜干燥。
[0083]
(10)小心加入15
‑
20μl depc水至滤膜中央,室温放置2min,全速离心1min,收 集rna。立即使用或放于
‑
80℃备用。
[0084]
2、将提取的rna反转录为cdna
[0085]
用hiscripttm q rt supermix for qpcr(+gdna wiper)(vazyme,china)试剂盒将 提取的rna反转录为cdna。
[0086]
3、ab
‑
atps表达量检测
[0087]
按照试剂盒sybr green real
‑
time pcr master mix
‑
plus kit说明书操作,以合成的相 应cdna为模板,分别测定卵、幼虫、雌虫和雄虫的ab
‑
atps表达量,共设三个重复。
[0088]
ab
‑
atps基因表达量检测使用引物如下:
[0089]
qd2f:5
’‑
acttcgaggacattctccgtt
‑3’
[0090]
qd2r:5
’‑
catgatctccggtcgaacctg
‑3’
[0091]
根据编码水稻干尖线虫rdna的18s rrna基因序列(ay508035)设计一对特异引物 ab18sf和ab18sr,进行扩增得到140bp片段作为内参基因,引物序列如下:
[0092]
ab18sf:5
’‑
ctcgtggtggctggtatgctg
‑3’
[0093]
ab18sr:5
’‑
gtttcccgtgttgagtcaaattaag
‑3’
[0094]
qpcr使用仪器为cfx
‑
96(bio
‑
rad),实时荧光定量的数据分析采用bio
‑
rad公司 提供的cfx manger软件分析。
[0095]
二、实验结果
[0096]
ab
‑
atps在水稻干尖线虫不同虫态中的表达量qpcr检测结果显示,ab
‑
atps的表达量 由高到低为幼虫、卵、雌虫和雄虫,前三个虫态表达量分别是雄虫的26.79、13.10和4.86 倍(图2)。除了雌虫和雄虫之间的表达量差异不显著(p>0.05),其他虫态之间的表达 量差异均显著(p<0.05)。
[0097]
实施例3 ab
‑
atps基因的原位杂交
[0098]
一、实验方法
[0099]
将约10000条混合虫态线虫液定容至30~50μl,添加3%的多聚甲醛溶液5℃固定
18 h,22℃固定4h。根据ab
‑
atps的cdna全长基因设计引物,并作为模板合成dig标记 的rna探针(roche,germany),具体步骤如下:
[0100]
1、rna探针体外转录模板(dna)的扩增引物
[0101]
本试验利用体外转录法合成ab
‑
atps的rna探针,体外转录rna探针所需的模板 dna通过pcr获得,模板dna的扩增引物设计如下:
[0102]
a.反义链体外转录模板的扩增引物:
[0103]
ia
‑
d2f:5
′‑
taatacgactcactatagggaaatacgcgaccagtttgtatcagg
‑3′
[0104]
ia
‑
d2r:5
′‑
ccgactgtttcaagttgcgat
‑3′
[0105]
b.正义链体外转录模板的扩增引物:
[0106]
is
‑
d2f:5
′‑
taatacgactcactatagggccgactgtttcaagttgcgat
‑3′
[0107]
is
‑
d2r:5
′‑
aaatacgcgaccagtttgtatcagg
‑3′
[0108]
2、rna探针体外转录模板(dna)的获得
[0109]
用含ab
‑
atps全长基因的重组质粒,作为扩增rna探针转录所需的dna模板。正义 链和反义链模板dna使用引物ia
‑
d2f和ia
‑
d2r扩增,pcr反应体系配制和反应条件 参照实例1进行。洗脱时用无rna酶的水溶解,确定其浓度,浓度需在0.5
‑
1μg/μl范围 内。获得的正反义链转录模板保存于4℃。
[0110]
3、rna探针的合成
[0111]
分别合成正义和反义rna探针,反应体系(共20μl)如下:
[0112][0113]
反应条件:37℃温育2h,电泳检验产物。
[0114]
4、探针的纯化
[0115]
制备好的探针加入10u dnase i,5μl 10
×
dnase i buffer,37℃水浴15min以去除 dna后进行纯化和浓缩,将rna探针添加至含线虫的杂交液。杂交完成后用微分干扰显 微镜检查及拍照。
[0116]
二、实验结果
[0117]
原位杂交的结果显示,ab
‑
atps mrna定位在水稻干尖线虫雌虫的食道以及生殖系统 (图3),正义探针杂交的线虫,没有检测到杂交信号。
[0118]
实施例4 ab
‑
atps基因rnai干扰
[0119]
一、实验方法
[0120]
ab
‑
atps基因的rnai采用双链rna(dsrna)浸泡的方法。双链rna的合成采用 体外转录的方法。ab
‑
atps基因的orf克隆至pmd18
‑
t载体中(takara,japan),克隆需 经测序验
证。参照script maxtm thermo t7 transcription kit(toyobo,japan)操作手 册体外转录合成用于沉默ab
‑
atps基因和对照gfp基因的dsrna,dsrna合成产物纯化后 用电泳检测完整性、nano
‑
drop spectrophotometer检测质量及浓度,
‑
80℃保存。具体步骤 如下:
[0121]
1、体外转录正、反义链单链rna(single strand rna,ssrna)的获得
[0122]
ab
‑
atps和gfp的ssrna合成方法按照实施例3的方法获得ab
‑
atps部分片段的正义 链和反义链的ssrna。gfp ssrna合成的模板为实验室保存的含gfp全长基因的质粒。
[0123]
gfp部分片段的正义链和反义链的ssrna合成引物序列如下:
[0124]
反义链模板引物:
[0125]
g
‑
t7a:5
′‑
ggatcctaatacgactcactataggg cgatgcggttcaccagggtgtcg
‑3′
[0126]
g
‑
s:5
′‑
cacaagttcagcgtgtccggcg
‑3′
[0127]
正义链模板引物:
[0128]
g
‑
t7s: 5
′‑
ggatcctaatacgactcactatagggcacaagttcagcgtgtccggcg
‑3′
[0129]
g
‑
a:5
′‑
cgatgcggttcaccagggtgtcg
‑3′
[0130]
2、体外转录合成双链rna(double strand rna,dsrna)
[0131]
(1)ssrna合成结束后加入5u的rq1 rnase
‑
free dnase,37℃条件反应15min以 消化反应液中的dna;
[0132]
(2)电泳检测ssrna的质量并检测浓度;
[0133]
(3)在depc处理的新的1.5ml的离心管中加入等比例的正义和反义链rna,轻 轻混合摇匀;
[0134]
(4)94℃孵育10min后,自然降温至室温退火形成dsrna;
[0135]
(5)使用移液器加入5μl rnaset1,37℃孵育30min。
[0136]
(6)制备好的探针加入10u dnase i,5μl 10
×
dnase i buffer,37℃水浴15min以 去除dna后进行纯化和浓缩,并检测其浓度,于
‑
80℃保存备用。
[0137]
3、dsrna浸泡处理
[0138]
分离收集胡萝卜愈伤组织上培养的水稻干尖线虫于depc处理过的离心管中,每个离 心管中收集混合虫态线虫500条,加入50μl ab
‑
atps dsrna(2μg/μl)浸泡,浸泡处理 时间分别为12h、24h、36h和48h,用50μl的gfp dsrna(2μg/μl)浸泡作为处理对 照,共8个处理,每个处理重复3次。浸泡在25℃下进行。
[0139]
4、浸泡处理后ab
‑
atps沉默效率检测
[0140]
将以上各处理线虫,用depc水清洗三次后提取rna,用qpcr仪测定ab
‑
atps表达 量,方法同实施例2。每个处理重复3次,每个重复样品技术重复2次,最终结果取6次 检测的平均值。
[0141]
二、实验方法
[0142]
得到的ab
‑
atps dsrna为由如seq id no.3所示的核苷酸序列作为正义链及由与seqid no.3所示的核苷酸序列反向互补的核苷酸序列作为反义链组成的双链rna。
[0143]
用ab
‑
atps dsrna浸泡水稻干尖线虫后,用qpcr对ab
‑
atps基因的沉默效率进行检 测。结果表明(图4),经ab
‑
atps dsrna浸泡处理12h、24h、36h和48h的线虫,ab
‑
atps 的相对表达量与对应的gfp dsrna浸泡相同时间的处理相比,分别下降了86.7%、80.7%、 86.3%和75.4%,差异显著(p<0.05);ab
‑
atps基因rnai处理的沉默效率随着时间延长 差异不显
著(p>0.05);对照组中gfp dsrna浸泡各处理时间的表达量差异不显著(p>0.05)。
[0144]
实施例4 ab
‑
atps基因与水稻osrlk3基因存在互作
[0145]
一、实验方法
[0146]
按照clonexpress ii one step cloning kit(vazyme,china)将ab
‑
atps基因全长构建 到载体pgadt7载体的ad结构域中。
[0147]
扩增引物序列为:
[0148]
add2f:5
’‑
cagctcgagctcgatggatcctccgcaagtcgtgccgcc
‑3’
[0149]
add2r:5
’‑
gccatggaggccagtgaattctgatggtctcgttgagtttcttga
‑3’
[0150]
反应体系:
[0151][0152]
反应条件:94℃反应2min;98℃反应10s,55℃反应30s,72℃反应15s,30个循 环;72℃反应10min。
[0153]
双酶切位点为ncoi/bamhi。序列确定无误后,连接以获得重组质粒,并转化到大肠 杆菌(escherichia coli)感受态细胞dh5α中;挑取阳性克隆测序(sangon biotech,china) 并提取重组质粒。参照matchmaker two
‑
hybrid system(clontech,usa)的说明书,将连 接好的载体转化至y187酵母感受态细胞中,随后与含osrlk3基因的y2h酵母感受态 进行小规模共转化实验,菌液全部涂布于qdo/xa/aba平板上,用封口膜封闭好,30℃ 倒置培养3
‑
5d,产生的蓝色菌落说明ad与bd存在互作关系。
[0154]
二、实验结果
[0155]
将基因ab
‑
atps编码区重新构建到gal4
‑
ad(pgadt7)结构域,与gal4
‑
bd(pgbkt7) 结构域为含去信号肽的osrlk3序列片段进行小规模共转化,使用qdo培养基进行阳性 克隆筛选,培养3~7d后,挑选单克隆印记于qdo/xa培养基上培养,48h内菌落呈现与 阳性对照一致的蓝色,而作为阴性对照的共转pgadt7空载体和pgbkt7
‑
osrlk3酵母, pgadt7
‑
ab
‑
atps和pgbkt7空载体酵母在qdo/xa培养基上并无生长。此结果说明osrlk3和ab
‑
atps之间存在互作(图5)。
[0156]
实施例5 ab
‑
atps基因沉默对水稻干尖线虫繁殖力的影响以及对水稻osrlk3基因表达量 的影响
[0157]
一、实验方法
[0158]
1、混合虫态的水稻干尖线虫分别用实施例3的ab
‑
atps dsrna浸泡处理12h、24h、 36h和48h,以实施例3的gfp dsrna处理12h、24h、36h和48h作为对照处理。每种 处理挑取30条雌虫接种于胡萝卜愈伤组织上,每个处理重复9次。接种后的胡萝卜愈伤 组织放在25℃培养箱黑暗培养35d,分离并统计胡萝卜愈伤组织上的线虫。
[0159]
2、混合虫态的水稻干尖线虫分别用实施例3的ab
‑
atps dsrna和gfp dsrna浸泡处 理48h,未接种水稻干尖线虫的水稻作为空白对照处理,分别取dai0.5、dai1、dai2、 dai3、
和dai7的水稻植株地上部位rna提取使用general rna extraction kit(dongsheng, china)进行,对照为同一时间点未接线虫的水稻(ck)用于检测水稻osrlk3基因的表 达量,qpcr以及数据分析方法同实施例2。每个处理设3个重复,每个重复样品由3株 水稻组成,每个样品pcr重复两次。
[0160]
二、实验结果
[0161]
用ab
‑
atps dsrna浸泡水稻干尖线虫后,用qpcr对ab
‑
atps基因的沉默效率进行检 测。结果表明(图6a),经ab
‑
atps dsrna浸泡处理12h、24h、36h和48h的线虫, ab
‑
atps的相对表达量与对应的gfp dsrna浸泡相同时间的处理相比,分别下降了86.7%、 80.7%、86.3%和75.4%,差异显著(p<0.05);ab
‑
atps基因rnai处理的沉默效率随着时 间延长差异不显著(p>0.05);对照组中gfp dsrna浸泡各处理时间的表达量差异不显著 (p>0.05)。经ab
‑
atps dsrna浸泡处理不同时间的水稻干尖线虫,在胡萝卜片上培养35 d后的繁殖结果表明(图6a):ab
‑
atps被rnai处理12h、24h、36h和48h的水稻干 尖线虫,其繁殖数量均显著低于(p<0.05)经gfp dsrna浸泡相同时间处理的对照组线虫, 并且随着浸泡时间的延长繁殖量不断降低,除了处理12h和24h之间以及36h和48h之 间的差异不显著外(p>0.05),其他处理之间的差异显著(p<0.05)。经gfp dsrna浸泡 的各处理线虫繁殖量随浸泡时间的增加,无明显差异(p>0.05),在48h内,未随处理时 间延长明显降低。
[0162]
选择ab
‑
atps dsrna和gfp dsrna浸泡48h后水稻干尖线虫接种水稻后,对水稻 osrlk3基因的表达量检测结果表明,osrlk3基因表达量,在dai 0.5ab
‑
atps dsrna处 理显著低于gfp dsrna处理(p<0.05),而在dai 1ab
‑
atps dsrna处理显著高于gfp dsrna 处理(p<0.05),在dai 2
‑
7则两处理无显著差异(p>0.05)(图6b)。ab
‑
atps dsrna 处理中,osrlk3表达量在dai1显著最高(p<0.05),gfp dsrna处理中,osrlk3表达 量在在dai 0.5表达量显著最高。因此,使用沉默ab
‑
atps基因后的水稻干尖线虫对水稻 osrlk3基因表达存在影响,也表明ab
‑
atps和osrlk3存在互作关系。
[0163]
实施例6 ab
‑
atps基因的瞬时表达
[0164]
一、实验方法
[0165]
1、植物表达载体构建
[0166]
使用的植物瞬时表达载体为pcambia1300,分别按照clonexpress ii one step cloning kit(vazyme,china)将ab
‑
atps和gfp基因全长连接到同一个pcambia1300载体上。 另使用同样方法单独构建只连接gfp基因的pcambia1300载体作为对照。具体步骤如下:
[0167]
pcambia1300
‑
gfp载体构建:
[0168]
引物序列:
[0169]
gf:5
’‑
cgcgtcgacatgtccgcaagtcgt
‑3’
[0170]
gr:5
’‑
tggctgcagttaaacggcctctttgat
‑3’
[0171]
pcr反应体系及条件参照实施例1。
[0172]
pcambia1300
‑
ab
‑
atps
‑
gfp载体构建引物:
[0173]
pd2f:5
’‑
atcgagctcatggtgagcaagggc
‑3’
[0174]
pd2r:5
’‑
cgcggatcccttgtacagctcgtc
‑3’
[0175]
pcr反应体系及条件参照实施例1。模板为ab
‑
atps基因全长序列片段。
[0176]
连接ab
‑
atps基因序列的双酶切位点为sali/psti;绿色荧光蛋白(gfp)基因序列
双酶 切位点为saci/bamhi;分别按照clonexpress ii one step cloning kit(vazyme,china)将 ab
‑
atps和gfp基因全长连接到同一个pcambia1300载体上。另使用同样方法单独构建 只连接gfp基因的pcambia1300载体作为对照,构建好的载体转化至大肠杆菌dh5α中,。 挑选阳性克隆测序验证后,收集菌液,提取质粒备用。
[0177]
2、农杆菌gv3101转化
[0178]
将连接好的载体转化到农杆菌gv3101感受态细胞中,具体步骤如下参见农杆菌感受 态细胞说明书。挑选阳性克隆经pcr验证后备用。鉴定正确的工程农杆菌菌液中加入等 倍体积的50%甘油,液氮冷冻后置于
‑
80℃保存。
[0179]
3、注射烟草
[0180]
挑选的阳性克隆,使用含50μg/ml rif和50μg/ml kan的lb液体培养基,在28℃,250rpm条件下培养24
‑
48h。6000rpm离心5min浓缩菌体,用10mm,ph=5.5的mes 清洗3次。然后用mes稀释菌体至od600=0.5,再加入终浓度为100μm的乙酰丁香酮, 28℃避光静置4h;用1m l注射器的针头在四周龄烟草的叶片上轻轻扎一盒小孔,然后 用去除针头的注射器取适量菌液在小孔处将菌液打进烟草叶片。每片叶子打4个小孔,分 别注射带有pcambia1300
‑
ab
‑
atps
‑
gfp、pcambia1300
‑
gfp、pcambia1300空载体 质粒的工程农杆菌和mes,观察叶片是否产生细胞性坏死并使用荧光显微镜观察基因表 达情况。
[0181]
4、烟草总蛋白提取
[0182]
(1)烟草叶片总蛋白提取按照植物蛋白质提取试剂盒(购自凯基生物有限公司)说 明书进行,具体步骤如下:
[0183]
(2)分别收集被注射带有pcambia1300
‑
ab
‑
atps
‑
gfp、pcambia1300
‑
gfp质粒 的工程农杆菌和mes的烟草叶盘,加入液氮充分研磨,避免反复冻融;
[0184]
(3)将研磨后的粉末转入1.5ml离心管中,加入300μl裂解液;
[0185]
(4)在上述液体中加入适量的蛋白酶抑制剂,置于冰上30min,其间将离心管反转 几次;
[0186]
(5)4℃,14000rpm离心20min,所得到的上清液即为烟草蛋白质溶液,将蛋白质 溶液保存于
‑
20℃备用。
[0187]
5、sds电泳
[0188]
6、western blot
[0189]
目的蛋白使用gfp抗体(购自北京全式金生物试剂有限公司)进行western blot检测, 具体步骤如下:
[0190]
(1)电泳结束后,将胶从玻璃板中轻轻取出,放入装有转膜液的容器中,浸泡10min;
[0191]
(2)将pvdf膜剪成名片大小,为防止手上的蛋白质成分污染,应使用手套。将pvdf 膜浸润在甲醇溶液中10
‑
30s,再将pvdf膜浸泡在蒸馏水中,振荡5min,最后将pvdf 膜浸泡在转膜液中,振荡10min;
[0192]
(3)将滤纸剪成名片大小,共需8张,放入转膜液中浸湿。从阴极开始,按照滤纸 4张
→
sds
‑
page凝胶
→
pvdf膜
→
滤纸4张
→
阳极的顺序叠放,避免凝胶与膜之间产生 气泡;
[0193]
(4)以pvdf膜的面积(cm2)
×
0.8ma的公式计算的电流强度,通电1h,转膜结 束后,将pvdf膜浸入盛有pbst的塑料容器内,振荡5min将膜洗净;
[0194]
(5)将pvdf膜浸入5%的脱脂奶粉溶液中,在室温条件下振荡1h;
[0195]
(6)按照1∶5000的比例用pbs稀释一抗,将pvdf膜浸入一抗稀释溶液中,在室 温下振荡2h使pvdf膜和一抗充分接触。
[0196]
(7)用pbst振荡洗涤3次,每次10min。
[0197]
(8)按照1∶5000的比例用pbs稀释二抗,将pvdf膜浸入二抗稀释溶液中,在室 温下振荡1h,使pvdf膜和二抗充分接触。
[0198]
(9)用pbst振荡洗涤3次,每次10min。
[0199]
(10)凝胶成像仪拍照。
[0200]
二、实验结果
[0201]
为进一步确认ab
‑
atps在植物细胞中的位置,将其与gfp进行融合,在烟草表皮细 胞中进行瞬时表达(图7)。在单独表达gfp蛋白和ab
‑
atps
‑
gfp融合表达的烟草细胞 中,荧光均分布于细胞核、细胞质和细胞膜。提取瞬时表达烟草叶片蛋白,用于western blot 检测,结果表明,gfp蛋白对照在约40kda处出现条带(大小约为37kda),ab
‑
atps
‑
gfp 蛋白条带出现在50
‑
70kda之间(大小约为57kda),说明ab
‑
atps
‑
gfp融合蛋白表达 正确。对瞬时表达叶片注射农杆菌后第5d,注射含pcambia1300
‑
ab
‑
atps
‑
gfp质粒农 杆菌位点出现细胞死亡,而注射含pcambia1300
‑
gfp质粒农杆菌位点、mes缓冲液和 pcambia1300空载体质粒农杆菌均无变化,由此认为ab
‑
atps蛋白可引发接种位点的细 胞凋亡(图7c)。
[0202]
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范 围的限制,对于本领域的普通技术人员来说,在上述说明及思路的基础上还可以做出其它 不同形式的变化或变动,这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神 和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围 之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1