含硝酸酯官能团的化合物及其合成方法
本发明属于有机合成化学技术领域,涉及一种含硝酸酯官能团的化合物及其合成方法。
背景技术:
过渡金属催化的c-h键官能化策略提供了一种简洁的途径,将c-h键直接转化为各种更具附加价值的碳-碳键和碳-杂原子键(例如,c-卤、c-o、c-n和c-s等),这将简化许多合成步骤并减少不需要的副产物的形成。体系中导向基团导向策略的构建使得反应的区域选择性和立体选择性也得到了很好的控制。硝酸酯类化合物在医药和含能材料领域有着重要的应用,如作为一般性冠心病、心绞痛治疗药物以及含能材料推进剂等。传统的硝酸酯类化合物的合成方法是使用浓硝酸/硫酸的混合物对醇类底物进行硝酸酯化,该反应条件苛刻,官能团耐受性差且对环境造成了污染。或者硝酸酯化反应位点为烯丙基位(chemicalandpharmaceuticalbulletin,1984,32(3):887-890)或苄基位(tetrahedronletters52(2011)4654-4657)等非惰性碳氢位点,适用底物范围特别有限。或者进行卤素取代反应(journalofmedicinalchemistry,2004,47(3):711-719)生成硝酸酯类结构,但需要活化安装其它官能团,反应步骤复杂。完全惰性的c(sp3)-h键直接特定位点选择性转化为硝酸酯基的反应策略未见报道。
技术实现要素:
本发明的目的之一在于提供一种含硝酸酯官能团的化合物,其化学结构式如下:
本发明的目的之二在于提供一种条件温和、较高产率、选择性好绿色环保的含硝酸酯官能团的化合物的合成方法,反应通式为:
肟喹啉导向基团修饰的醇底物在乙酸钯的催化作用下,以亚硝酸银为硝酸酯源,选择性氟试剂(selectfluor)为氧化剂,四丁基硫酸氢铵为相转移催化剂,1,2-二氯乙烷为溶剂,采用一锅法,在70~90℃下反应12~24h,反应结束后过短硅胶柱,旋干移除溶剂,水洗萃取,除去溶剂得粗产物,粗产物经柱层析分离后即得含硝酸酯官能团的化合物,所述的导向基团修饰的醇分子的结构式为:
优选的,乙酸钯摩尔量为醇底物摩尔量的0.05-0.2equive,更优选为0.1equive。
优选的,亚硝酸银摩尔量为醇底物摩尔量的1.5-4.0equive,更优选为3.0equive。
优选的,选择性氟试剂摩尔量为醇底物摩尔量的1.0-2.0equive,更优选为1.5equive。
优选的,四丁基硫酸氢铵摩尔量为醇底物摩尔量的0.5-1.0equive,更优选为1.0equive。
优选的,反应温度为80℃,反应时间为18h。
优选的,柱层析中采用的洗脱剂为乙酸乙酯:石油醚的体积比=1:10的混合溶液。
与现有技术相比,本发明的优点如下:
(1)采用广泛易得、价格便宜的小分子醇为起始原料,首次实现选择性活化醇β-c(sp3)-h合成c-ono2键新型化合物,弥补了现有碳氢键官能团化硝酸酯化方法上的空缺,且底物适用性广泛。
(2)运用碳氢活化的反应方法,避免了常规的合成方法的预活化,原子经济性高,方法绿色简便,产率良好至优异。
(3)导向基团易于安装和脱除,便于硝酸酯化产物的实际应用。
附图说明
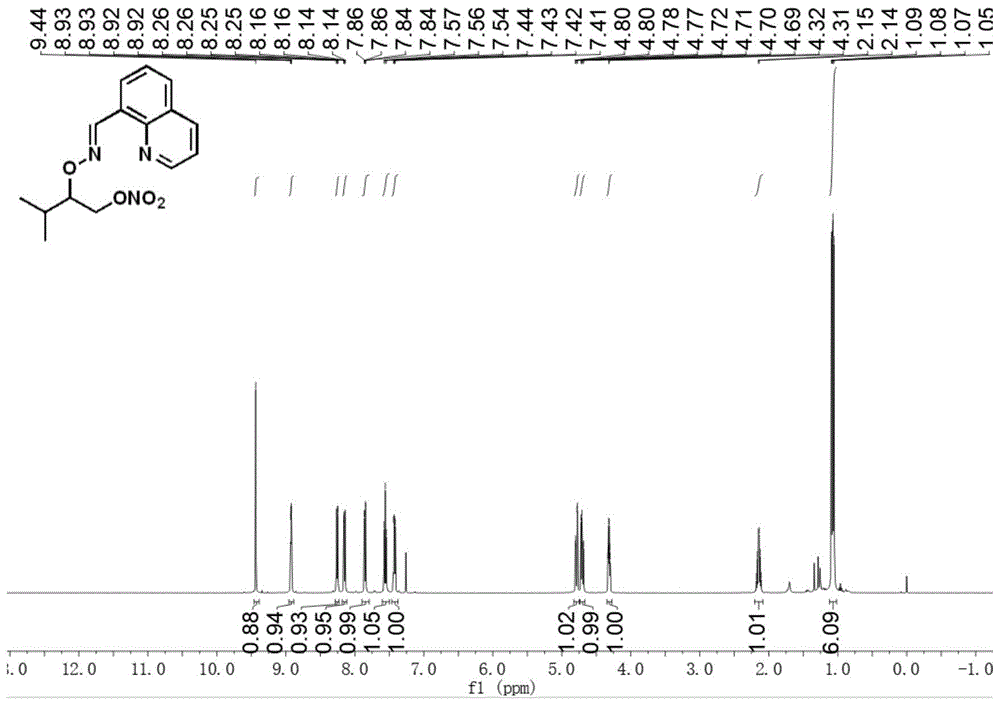
图1为实施例1中产物的核磁共振氢谱(1hnmr)。
图2为实施例1中产物的核磁共振碳谱(13cnmr)。
图3为实施例2中产物的核磁共振氢谱(1hnmr)。
图4为实施例2中产物的核磁共振碳谱(13cnmr)。
图5为实施例3中产物的核磁共振氢谱(1hnmr)。
图6为实施例3中产物的核磁共振碳谱(13cnmr)。
图7为实施例4中产物的核磁共振氢谱(1hnmr)。
图8为实施例4中产物的核磁共振碳谱(13cnmr)。
具体实施方式
下面通过实施例和附图对本发明作进一步详细说明。肟喹啉导向基团修饰的醇底物的制备参考文献【naturechemistry.2015,7(10),829-34】。
本发明是在乙酸钯的催化作用下,以亚硝酸银为硝酸酯化试剂,以选择性氟试剂为氧化剂,四丁基硫酸氢铵为相转移催化剂,1,2-二氯乙烷为溶剂,将安装过导向基团的醇类底物加入其中,反应在空气氛围下加热至70~90℃进行,反应结束后,过柱纯化以良好的产率得到β位硝酸酯化的产物。本发明方法条件温和,能够有效地一步构建碳-氧原子化学键,省去了预官能团化过程,减少了反应步骤,具有较高的原子经济性。同时底物适用范围广,对简单的醇类小分子结构进行修饰,生成含硝酸酯官能团的新型化合物,该化合物在医药和含能材料等领域具有较高的潜在价值和广阔的应用前景。
本发明实现了过渡金属钯催化肟喹啉导向基团导向的醇类化合物选择性β-c(sp3)-h硝酸酯化官能团化,丰富现有对醇类化合物的结构修饰策略,完善官能团化方法体系的构建。本发明涉及的反应有自由基的参与,在加热条件和氧化剂作用下激发自由基,自由基转化为硝酸酯结构加成到钯环中间体,最终经过还原消除过程得到产物及二价钯催化剂,完成催化循环。
实施例1
e)-3-甲基-2-(((喹啉-8-亚甲基)氨基)氧)硝酸丁酯
准确称量e)-喹啉甲乙醛-(3-甲基丁烷-2-酰基)肟(24.2mg,0.1mmol),乙酸钯(2.2mg,0.01mmol),亚硝酸银(46.2mg,0.3mmol),选择性氟试剂(53.1mg,0.15mmol),四丁基硫酸氢铵(33.9mg,0.1mmol)转移至反应容器中,加入1.5ml1,2-二氯乙烷,旋紧瓶塞,80℃反应18h。反应结束后将反应液冷却至室温,短硅胶过滤除去难溶杂质,用乙酸乙酯和水萃取3次,用卤水萃取1次,有机相去除溶剂得粗产物,粗产物进行柱层析分离(洗脱剂:乙酸乙酯/石油醚=1:10),得到纯净干燥的产物,产率72%。1hnmr(500mhz,cdcl3)δ9.44(s,1h),8.92(dd,j=4.1,1.7hz,1h),8.26(dd,j=7.3,0.8hz,1h),8.15(dd,j=8.3,1.6hz,1h),7.85(dd,j=8.1,0.9hz,1h),7.56(t,j=7.7hz,1h),7.43(dd,j=8.3,4.2hz,1h),4.79(dd,j=11.9,3.5hz,1h),4.70(dd,j=11.9,6.5hz,1h),4.32(td,j=6.4,3.6hz,1h),2.15(dq,j=13.6,6.8hz,1h),1.07(dd,j=11.0,6.9hz,6h).13cnmr(126mhz,cdcl3)δ174.70,150.06,146.69,145.87,136.27,129.82,129.69,128.31,126.40,126.22,121.46,84.08,72.57,29.42,18.54,18.22.
实施例2
e)-2-环己基-2-(((喹啉-8-亚甲基)氨基)氧)硝酸乙酯
准确称量e)-喹啉-8-甲醛o-(1-环己基乙基)肟(28.2mg,0.1mmol),乙酸钯(2.2mg,0.01mmol),亚硝酸银(46.2mg,0.3mmol),选择性氟试剂(53.1mg,0.15mmol),四丁基硫酸氢铵(33.9mg,0.1mmol)转移至反应容器中,加入1.5ml1,2-二氯乙烷,旋紧瓶塞,80℃反应18h。反应结束后将反应液冷却至室温,短硅胶过滤除去难溶杂质,用乙酸乙酯和水萃取3次,用卤水萃取1次,有机相去除溶剂得粗产物,粗产物进行柱层析分离(洗脱剂:乙酸乙酯/石油醚=1:10),得到纯净干燥的产物,产率63%。1hnmr(500mhz,chloroform-d)δ9.42(s,1h),8.94(dd,j=4.1,1.8hz,1h),8.26(dd,j=7.3,1.3hz,1h),8.17(dd,j=8.3,1.8hz,1h),7.86(dd,j=8.1,1.4hz,1h),7.57(t,j=7.7hz,1h),7.44(dd,j=8.3,4.2hz,1h),4.81(dd,j=11.9,3.6hz,1h),4.70(dd,j=11.8,6.4hz,1h),4.33(td,j=6.5,3.5hz,1h),1.96(d,j=13.0hz,1h),1.79(dt,j=12.3,3.7hz,4h),1.70(d,j=12.7hz,2h),1.20(ddd,j=11.8,5.4,2.8hz,1h).13cnmr(126mhz,chloroform-d)δ150.05,146.60,145.84,136.30,129.82,129.66,128.30,126.41,126.26,121.46,83.57,72.53,38.98,28.93,28.60,26.34,26.09,25.97.
实施例3
e)-4-苯基-2-(((喹啉-8-亚甲基)氨基)氧)硝酸丁酯
准确称量e)-喹啉-8-碳醛o-(4-苯基丁烷-2-酰基)肟(30.4mg,0.1mmol),乙酸钯(2.2mg,0.01mmol),亚硝酸银(46.2mg,0.3mmol),选择性氟试剂(53.1mg,0.15mmol),四丁基硫酸氢铵(33.9mg,0.1mmol)转移至反应容器中,加入1.5ml1,2-二氯乙烷,旋紧瓶塞,80℃反应18h。反应结束后将反应液冷却至室温,短硅胶过滤除去难溶杂质,用乙酸乙酯和水萃取3次,用卤水萃取1次,有机相去除溶剂得粗产物,粗产物进行柱层析分离(洗脱剂:乙酸乙酯/石油醚=1:10),得到纯净干燥的产物,产率67%。1hnmr(500mhz,cdcl3)δ9.50(s,1h),8.95(dd,j=4.1,1.7hz,1h),8.31–8.24(m,1h),8.17(dd,j=8.3,1.5hz,1h),7.88(dd,j=8.1,0.8hz,1h),7.59(t,j=7.7hz,1h),7.45(dd,j=8.3,4.2hz,1h),7.31(t,j=7.5hz,2h),7.23(dd,j=18.2,7.2hz,3h),4.77–4.67(m,2h),4.56(dq,j=9.3,4.7hz,1h),2.97–2.86(m,1h),2.85–2.73(m,1h),2.26–2.12(m,1h),2.00(dddd,j=11.4,9.7,7.0,4.5hz,1h).13cnmr(126mhz,cdcl3)δ150.15,147.24,145.88,141.21,136.35,129.86,129.72,128.56,128.34,126.43,126.36,126.16,121.54,78.49,73.75,32.44,31.48.
实施例4
(e)-4-(硝基氧基)-3-(((喹啉-8-基亚甲基)氨基)氧基)3-甲氧基苯甲酸丁酯
准确称量(e)-3-(((喹啉-8-亚甲基)氨基)氧基)3-甲氧基苯甲酸丁酯(31.2mg,0.1mmol),乙酸钯(2.2mg,0.01mmol),亚硝酸银(46.2mg,0.3mmol),选择性氟试剂(53.1mg,0.15mmol),四丁基硫酸氢铵(33.9mg,0.1mmol)转移至反应容器中,加入1.5ml1,2-二氯乙烷,旋紧瓶塞,80℃反应18h。反应结束后将反应液冷却至室温,短硅胶过滤除去难溶杂质,用乙酸乙酯和水萃取3次,用卤水萃取1次,有机相去除溶剂得粗产物,粗产物进行柱层析分离(洗脱剂:乙酸乙酯/石油醚=1:10),得到纯净干燥的产物,产率71%。1hnmr(500mhz,cdcl3)δ9.45(s,1h),8.94(dd,j=4.2,1.7hz,1h),8.23(dd,j=7.4,1.3hz,1h),8.17(dd,j=8.4,1.7hz,1h),7.88(dd,j=8.2,1.3hz,1h),7.64(d,j=7.6hz,1h),7.59–7.54(m,2h),7.45(dd,j=8.3,4.2hz,1h),7.34(t,j=7.9hz,1h),7.09(dd,j=8.3,2.7hz,1h),4.85–4.76(m,3h),4.61–4.50(m,2h),3.84(s,3h),2.30(dd,j=9.7,4.1hz,1h),2.27–2.18(m,1h).13cnmr(126mhz,chloroform-d)δ166.34,159.59,150.14,147.67,145.82,136.31,131.36,129.95,129.47,128.29,126.37(d,j=4.4hz),122.02,121.54,119.61,114.04,76.51,73.43,61.21,55.46,30.12.
对比例1
当过渡金属催化剂选用二乙腈氯化钯时,产率为20%;选用氯化钯时,观测不到目标产物。
对比例2
当硝酸酯基源试剂选用tbn时,只能观察到微量产物;选用硝酸银时,产率为34%。
对比例3
当氧化剂选用三氟乙酸银时,产率为44%;选用过硫酸钾时,产率为10%。
对比例4
当不添加相转移催化剂时,目标产物的量为58%;选用氯化铵作相转移催化剂时,产率反而降至45%。
对比例5
当溶剂选用二氯甲烷时,产率为37%;选用氯仿时,产率为18%。
- 还没有人留言评论。精彩留言会获得点赞!