1.本发明属于医药生物技术领域,具体涉及一种单唾液酸四己糖神经节苷脂钠的精制方法。
背景技术:2.单唾液酸四己糖神经节苷脂(monosialotetrahexosylganglioside,gm1)是一种含唾液酸的糖神经鞘脂类物质,是已知的唯一能穿透血脑屏障的神经节苷脂。广泛存在于哺乳类动物的细胞膜上,尤以神经系统含量最为丰富,镶嵌于细胞双层脂质膜结构中,是神经细胞的重要组成成分之一,约占总脂质的10%。gm1在人体神经系统发生、生长、分化和再生过程中起着必不可少的作用。多年的实验及临床报道显示,gm1可促进受损神经恢复,具有促进神经重塑作用,是参与神经病变修复的主要物质之一。
3.现有技术中制备药用gm1可从哺乳动物脑中提取,具有积极的工业价值。不同哺乳动物来源的脑组织中gm1的含量有差异,但有资料显示猪脑组织提取的gm1与人源性gm1在结构和组成,特别是唾液酸的类型等方面完全一致。因此,从动物脑组织作为原料提取gm1,不会存在免疫原性和异常毒性反应。根据文献报道,牛脑中gm1含量相对其它牲畜较高,但鉴于牛源性病毒,如疯牛病等对人体健康的严重危害,一般选用相对较安全的猪脑为原料。
4.单唾液酸四己糖神经节苷脂钠的制法一般分为提取和纯化两个步骤;如中国专利申请cn103524572a公开了一种高纯度的单唾液酸四己糖神经节苷脂钠的制法,是以新鲜猪脑为原料经过提取步骤和纯化步骤进行制备,其中提取步骤包括制丙酮粉步骤、组织总脂的抽提步骤、folch分配步骤、脱病毒步骤、酸水解病毒灭活步骤;纯化步骤包括脱盐步骤、硅胶层析步骤、反相层析步骤、浓缩步骤、丙酮重结晶步骤、冻干步骤。中国专利申请cn102731584a中公开一种制备高纯度单唾液酸四己糖神经节苷脂(gm1)的方法,具体包括猪脑丙酮粉的制备、总神经节苷脂浓缩液的制备和酸水解步骤。
5.现有技术的制备方法中虽然取得了一定的技术效果,但仍存在或多或少的质量问题。由于临床上使用的单唾液酸四己糖神经节苷脂钠对人体具有较大的影响,使用纯度要求较高,因此,对单唾液酸四己糖神经节苷脂钠精制技术的改进具有重要的意义。
技术实现要素:6.为克服以上技术问题,本发明提供了一种单唾液酸四己糖神经节苷脂钠的精制方法。制成的单唾液酸四己糖神经节苷脂钠具有较高的纯度,产品质量较高,适用于推广应用。
7.为实现以上目的,本发明提供的技术方案如下:
8.一种单唾液酸四己糖神经节苷脂钠的精制方法,包括以下步骤:
9.(1)神经节苷脂猪脑提取物经正向不定型硅胶首次纯化后,后经辛烷基键合硅胶反相填料分离得神经节苷脂粗品;
10.(2)神经节苷脂粗品使用阳离子树脂进行离子交换,使用10k中空纤维超滤膜超滤
进行精制,得到高纯度单唾液酸四己糖神经节苷脂钠。
11.优选地,所述单唾液酸四己糖神经节苷脂钠的精制方法,包括以下步骤:
12.(1)猪脑经提取、萃取、水解后得猪脑提取物,备用;
13.(2)取无定形硅胶,用甲醇匀浆后装柱,使用三氯甲烷、甲醇和水的混合液做流动相进行平衡,取猪脑提取物用水加热溶解,用流动相进行洗脱,收集洗脱液,将洗脱液减压浓缩至粘稠,加入丙酮,静置沉淀,得到神经节苷脂粗品1;
14.(3)神经节苷脂粗品1溶解后,使用高压动态轴向压缩柱反相填料,以三氟乙酸甲醇溶液和水的混合溶液进行洗脱,收集洗脱液,并对洗脱液依据hplc高效液相色谱法进行检测;将洗脱液合并后减压浓缩至粘稠,加入丙酮,静置沉淀,得到神经节苷脂粗品2;
15.(4)粗品2加纯化水溶解后,加入活性炭,搅拌后,过滤,浓缩,加入丙酮沉淀,得神经节苷脂粗品3;
16.(5)用阳离子树脂装柱后,使用盐酸水溶液冲洗阳离子树脂柱,将神经节苷脂粗品3溶解上样,使用盐酸水溶液进行洗脱,收集洗脱液,超滤,至检测超滤液电导率≤10us/cm时停止超滤,得超滤液,将超滤液减压浓缩至粘稠液后加入丙酮,静置沉淀,过滤,干燥,得到高纯度单唾液酸四己糖神经节苷脂钠。
17.优选地,步骤(2)中,所述无定型硅胶的目数为200
‑
300目;
18.优选地,步骤(2)中,所述三氯甲烷、甲醇和水的体积比为65
‑
68:30
‑
33:3
‑
6;
19.优选地,步骤(2)中,所述三氯甲烷、甲醇和水的体积比为68:33:6;
20.优选地,步骤(2)中,所述加热的温度为30
‑
60℃;
21.优选地,步骤(2)中,所述加热的温度为50℃;
22.优选地,步骤(2)中,所述猪脑提取物与水加入比例为水:猪脑提取物=1l:80
‑
120g(v:w);
23.优选地,步骤(2)中,所述猪脑提取物与水加入比例为水:猪脑提取物=1l:100g(v:w);
24.优选地,步骤(2)中,所述流动相进行洗脱以20
‑
60ml/min的流速进行;
25.优选地,步骤(2)中,所述流动相进行洗脱以40ml/min的流速进行;
26.优选地,步骤(2)中,所述加入丙酮的体积为浓缩液体积的5
‑
10倍。
27.优选地,步骤(2)中,所述加入丙酮的体积为浓缩液体积的10倍。
28.优选地,步骤(3)中,所述反相填料为辛烷基键合硅胶反相填料粒径为10μm,孔径为0.5%三氟乙酸甲醇溶液和水的体积比为(68
‑
70):(30
‑
32)的流动相,柱规格为50
×
1000mm,紫外检测波长为205nm;
29.优选地,步骤(3)中,所述洗脱以60
‑
100ml/min的流速进行;
30.优选地,步骤(3)中,所述洗脱以80ml/min的流速进行;
31.优选地,步骤(3)中,所述高效液相色谱法进行检测的色谱条件为用十八烷基硅烷键合硅胶为填充剂,以0.01mol/l磷酸二氢钾溶液
‑
乙腈(32:68)为流动相a,以乙腈为流动相b;流速为每分钟1.2ml;检测波长为205nm;
32.优选地,步骤(3)中,所述加入丙酮的体积为浓缩液体积的5
‑
10倍。
33.优选地,步骤(3)中,所述丙酮的体积为浓缩液体积的10倍;
34.优选地,步骤(4)中,所述活性炭为针用活性炭,加入量为粗品2质量的3
‑
5%。
35.优选地,步骤(4)中,所述活性炭为针用活性炭,加入量为粗品2质量的5%。
36.优选地,步骤(5)中,所述盐酸水溶液为盐酸:纯化水=1l:5
‑
10l;
37.优选地,步骤(5)中,所述盐酸水溶液为盐酸:纯化水=1l:5l;
38.优选地,步骤(5)中,所述使用盐酸水溶液冲洗阳离子树脂柱以30
‑
70ml/min的流速进行1
‑
3h;
39.优选地,步骤(5)中,所述使用盐酸水溶液冲洗阳离子树脂柱以50ml/min的流速进行1h;
40.优选地,步骤(5)中,所述超滤使用10k中空纤维超滤膜进行;
41.优选地,步骤(5)中,所述加入丙酮的体积为粘稠液体积的5
‑
10倍。
42.优选地,步骤(5)中,所述加入丙酮的体积为粘稠液体积的10倍。
43.与现有技术比,本发明的技术优势在于:
44.(1)本方法先用200
‑
300目无定形硅胶把猪脑提取物进行首次纯化,得到神经节苷脂混合组分(含混合gm1组分约90%),采用tlc控制方法,后采用高压动态轴向压缩柱、辛烷基键合硅胶反相填料、三氟乙酸甲醇溶液和水的的流动相的精纯技术,可分别得到单唾液酸四己糖神经节苷脂gm1a和gm1b组分,后进行混合。可有效的分离去除的gma和gm1b之间包合的致敏杂质,该方法分离度高,可得到高纯度(99%)单唾液酸四己糖神经节苷脂。
45.(2)本发明提供的精制方法生产周期短、生产过程快速、成本低,易大工业生产操作。
附图说明
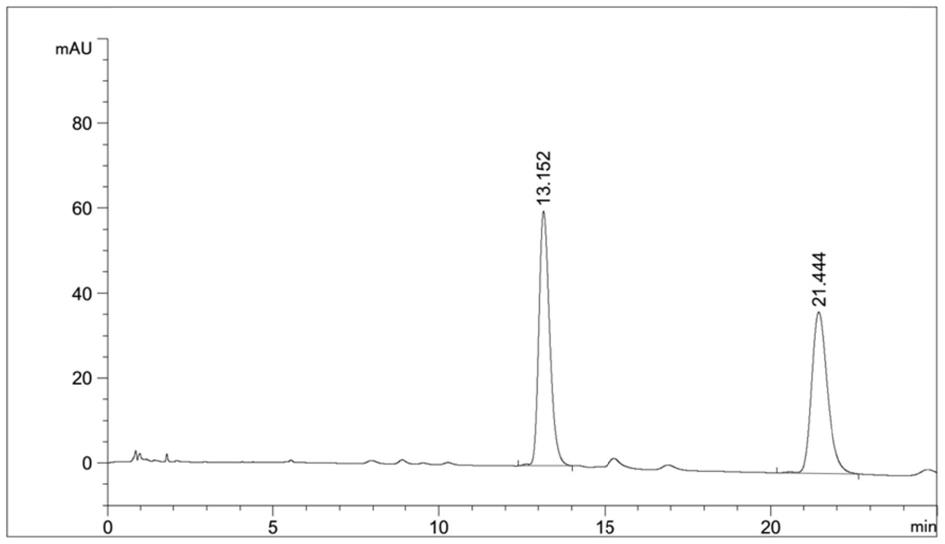
46.图1:实施例1所得产品的hplc谱图;
47.图2:实施例2所得产品的hplc谱图;
48.图3:实施例3所得产品的hplc谱图;
49.图4:实施例4所得产品的hplc谱图;
50.图5:实施例5所得产品的hplc谱图;
51.现结合附图和实施例对本发明作进一步说明。
具体实施方式
52.下面通过具体实施例对本发明进行说明,以使本发明技术方案更易于理解、掌握,但本发明并不局限于此。下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
53.实施例1
54.(1)将猪脑经提取、萃取、水解后得猪脑提取物,备用;
55.(2)取200目无定形硅胶2kg,6l甲醇匀浆后装柱(柱规格为100
×
800mm),使用三氯甲烷、甲醇和水的体积比为68∶33∶6的流动相进行平衡,取猪脑提取物用水加热50℃溶解,所述猪脑提取物与水加入比例约为水:猪脑提取物=1l:100g(v:w),用此流动相以40ml/min的流速进行洗脱,收集洗脱液,每2l收集一桶,依据tlc检测结果分阶段对洗脱液进行收集,将洗脱液减压浓缩至粘稠,加入浓缩液体积10倍量的丙酮,静置沉淀,得到神经节苷脂粗品1;
56.(3)将神经节苷脂粗品1溶解后,使用高压动态轴向压缩柱、反相填料(辛烷基键合硅胶反相填料粒径为10μm,孔径为0.5%三氟乙酸甲醇溶液和水的体积比为70∶30的流动相,柱规格为50
×
1000mm,紫外检测波长为205nm),以80ml/min的流速进行洗脱,收集洗脱液,每1000ml收集一瓶,分阶段收集gm1a和gm1b的洗脱液(如图1所示第一个出峰位置收集的为gm1a,第二个出峰位置为gm1b,以下实施例均同样处理),并对洗脱液依据hplc高效液相色谱法(色谱条件:用十八烷基硅烷键合硅胶为填充剂,以0.01mol/l磷酸二氢钾溶液
‑
乙腈(32:68)为流动相a,以乙腈为流动相b;流速为每分钟1.2ml;检测波长为205nm进行检测),将gm1a、gm1b洗脱液合并后减压浓缩至粘稠,加入浓缩液体积10倍量的丙酮,静置沉淀,静置沉淀,得到神经节苷脂粗品2;
57.(4)粗品2加纯化水溶解后,加入5%(m/m)的针用活性炭,搅拌30min后,过滤,浓缩,加入丙酮沉淀,得神经节苷脂粗品3;
58.(5)用001
×
7强酸型阳离子树脂装柱(柱规格为50
×
300mm)后,使用盐酸水溶液(盐酸:纯化水=1l:5l)以50ml/min的流速冲洗阳离子交换柱1小时,将神经节苷脂粗品3溶解上样,使用盐酸水溶液进行洗脱,收集洗脱液,然后用10k中空纤维超滤膜超滤,至检测超滤液电导率≤10us/cm时停止超滤,得超滤液,将超滤液减压浓缩至粘稠后加入10倍量的丙酮,静置沉淀,将沉淀液过滤,干燥,得到本发明高纯度单唾液酸四己糖神经节苷脂钠。经测定,单唾液酸四己糖神经节苷脂钠的hplc纯度为99.8%。其所得产品的hplc谱图如图1所示。
59.实施例2
60.(1)猪脑经提取、萃取、水解后得猪脑提取物,备用
61.(2)取300目无定形硅胶2kg,6l甲醇匀浆后装柱(柱规格为100
×
800mm),使用三氯甲烷、甲醇和水的体积比为68∶33∶6的流动相进行平衡,取猪脑提取物用水加热50℃溶解,所述猪脑提取物与水加入比例约为水:猪脑提取物=1l:100g(v:w),用此流动相以40ml/min的流速进行洗脱,收集洗脱液,每2l收集一桶,依据tlc检测结果分阶段对洗脱液进行收集,将洗脱液减压浓缩至粘稠,加入浓缩液体积10倍量的丙酮,静置沉淀,得到神经节苷脂粗品1;
62.(3)神经节苷脂粗品1溶解后,使用高压动态轴向压缩柱、反相填料(辛烷基键合硅胶反相填料粒径为10μm,孔径为0.5%三氟乙酸甲醇溶液和水的体积比为75∶25的流动相,柱规格为50
×
1000mm,紫外检测波长为205nm),以80ml/min的流速进行洗脱,收集洗脱液,每1000ml收集一瓶,分阶段收集gm1a和gm1b的洗脱液,并对洗脱液依据hplc高效液相色谱法(色谱条件:用十八烷基硅烷键合硅胶为填充剂,以0.01mol/l磷酸二氢钾溶液
‑
乙腈(32:68)为流动相a,以乙腈为流动相b;流速为每分钟1.2ml;检测波长为205nm)进行检测,将gm1a、gm1b洗脱液合并后减压浓缩至粘稠,加入浓缩液体积10倍量的丙酮,静置沉淀,静置沉淀,得到神经节苷脂粗品2;
63.(4)将粗品2加纯化水溶解后,加入5%(m/m)的针用活性炭,搅拌30min后,过滤,浓缩,加入丙酮沉淀,得神经节苷脂粗品3;
64.(3)用001
×
7强酸型阳离子树脂装柱(柱规格为50
×
300mm)后,使用盐酸水溶液(盐酸:纯化水=1l:5l)以50ml/min的流速冲洗阳离子交换柱1小时,将神经节苷脂粗品3溶解上样,使用盐酸水溶液进行洗脱,收集洗脱液,然后用10k中空纤维超滤膜超滤,至检测超
滤液电导率≤10us/cm时停止超滤,得超滤液,将超滤液减压浓缩至粘稠后加入10倍量的丙酮,静置沉淀,将沉淀液过滤,干燥,得到本发明高纯度单唾液酸四己糖神经节苷脂钠。经测定,单唾液酸四己糖神经节苷脂钠的hplc纯度为99.0%。其所得产品的hplc谱图如图2所示。
65.实施例3
66.(1)猪脑经提取、萃取、水解后得猪脑提取物,备用;
67.(2)取200目无定形硅胶2kg,6l甲醇匀浆后装柱(柱规格为100
×
800mm),使用三氯甲烷、甲醇和水的体积比为68∶33∶6的流动相进行平衡,取猪脑提取物用水加热50℃溶解,所述猪脑提取物与水加入比例约为水:猪脑提取物=1l:100g(v:w),用此流动相以40ml/min的流速进行洗脱,收集洗脱液,每2l收集一桶,依据tlc检测结果分阶段对洗脱液进行收集,将洗脱液减压浓缩至粘稠,加入浓缩液体积10倍量的丙酮,静置沉淀,得到神经节苷脂粗品1;
68.(3)神经节苷脂粗品1溶解后,使用高压动态轴向压缩柱、反相填料(辛烷基键合硅胶反相填料粒径为10μm,孔径为0.5%三氟乙酸甲醇溶液和水的体积比为68∶32的流动相,柱规格为50
×
1000mm,紫外检测波长为205nm),以80ml/min的流速进行洗脱,收集洗脱液,每1000ml收集一瓶,分阶段收集gm1a和gm1b的洗脱液,并对洗脱液依据hplc高效液相色谱法(色谱条件:用十八烷基硅烷键合硅胶为填充剂,以0.01mol/l磷酸二氢钾溶液
‑
乙腈(32:68)为流动相a,以乙腈为流动相b;流速为每分钟1.2ml;检测波长为205nm)进行检测,将gm1a、gm1b洗脱液合并后减压浓缩至粘稠,加入浓缩液体积10倍量的丙酮,静置沉淀,静置沉淀,得到神经节苷脂粗品2;
69.(4)粗品2加纯化水溶解后,加入5%(m/m)的针用活性炭,搅拌30min后,过滤,浓缩,加入丙酮沉淀,得神经节苷脂粗品3;
70.(5)将001
×
7强酸型阳离子树脂装柱(柱规格为50
×
300mm)后,使用盐酸水溶液(盐酸:纯化水=1l:5l)以50ml/min的流速冲洗阳离子交换柱1小时,将神经节苷脂粗品3溶解上样,使用盐酸水溶液进行洗脱,收集洗脱液,然后用10k中空纤维超滤膜超滤,至检测超滤液电导率≤10us/cm时停止超滤,得超滤液,将超滤液减压浓缩至粘稠后加入10倍量的丙酮,静置沉淀,将沉淀液过滤,干燥,得到本发明高纯度单唾液酸四己糖神经节苷脂钠。经测定,单唾液酸四己糖神经节苷脂钠的hplc纯度为99.2%。其所得产品的hplc谱图如图3所示。
71.实施例4
72.(1)猪脑经提取、萃取、水解后得猪脑提取物,备用
73.(2)取300目无定形硅胶2kg,6l甲醇匀浆后装柱(柱规格为100
×
800mm),使用三氯甲烷、甲醇和水的体积比为65:30:3的流动相进行平衡,取猪脑提取物用水加热30℃溶解,所述猪脑提取物与水加入比例约为水:猪脑提取物=1l:80g(v:w),用此流动相以20ml/min的流速进行洗脱,收集洗脱液,每2l收集一桶,依据tlc检测结果分阶段对洗脱液进行收集,将洗脱液减压浓缩至粘稠,加入浓缩液体积5倍量的丙酮,静置沉淀,得到神经节苷脂粗品1;
74.(3)神经节苷脂粗品1溶解后,使用高压动态轴向压缩柱、反相填料(辛烷基键合硅胶反相填料粒径为10μm,孔径为0.5%三氟乙酸甲醇溶液和水的体积比为70∶30的流
动相,柱规格为50
×
1000mm,紫外检测波长为205nm),以60ml/min的流速进行洗脱,收集洗脱液,每1000ml收集一瓶,分阶段收集gm1a和gm1b的洗脱液,并对洗脱液依据hplc高效液相色谱法(色谱条件:用十八烷基硅烷键合硅胶为填充剂,以0.01mol/l磷酸二氢钾溶液
‑
乙腈(32:68)为流动相a,以乙腈为流动相b;流速为每分钟1.2ml;检测波长为205nm)进行检测,将gm1a、gm1b洗脱液合并后减压浓缩至粘稠,加入浓缩液体积5倍量的丙酮,静置沉淀,静置沉淀,得到神经节苷脂粗品2;
75.(4)将粗品2加纯化水溶解后,加入3%(m/m)的针用活性炭,搅拌30min后,过滤,浓缩,加入丙酮沉淀,得神经节苷脂粗品3;
76.(3)将001
×
7强酸型阳离子树脂装柱(柱规格为50
×
300mm)后,使用盐酸水溶液(盐酸:纯化水=1l:10l)以30ml/min的流速冲洗阳离子交换柱3小时,将神经节苷脂粗品3溶解上样,使用盐酸水溶液进行洗脱,收集洗脱液,然后用10k中空纤维超滤膜超滤,至检测超滤液电导率≤10us/cm时停止超滤,得超滤液,将超滤液减压浓缩至粘稠后加入5倍量的丙酮,静置沉淀,将沉淀液过滤,干燥,得到本发明高纯度单唾液酸四己糖神经节苷脂钠。经测定,单唾液酸四己糖神经节苷脂钠的hplc纯度为99.6%。其所得产品的hplc谱图如图4所示。
77.实施例5
78.(1)猪脑经提取、萃取、水解后得猪脑提取物,备用
79.(2)取300目无定形硅胶2kg,6l甲醇匀浆后装柱(柱规格为100
×
800mm),使用三氯甲烷、甲醇和水的体积比为68:30:3的流动相进行平衡,取猪脑提取物用水加热60℃溶解,所述猪脑提取物与水加入比例约为水:猪脑提取物=1l:120g(v:w),用此流动相以60ml/min的流速进行洗脱,收集洗脱液,每2l收集一桶,依据tlc检测结果分阶段对洗脱液进行收集,将洗脱液减压浓缩至粘稠,加入浓缩液体积5倍量的丙酮,静置沉淀,得到神经节苷脂粗品1;
80.(3)神经节苷脂粗品1溶解后,使用高压动态轴向压缩柱、反相填料(辛烷基键合硅胶反相填料粒径为10μm,孔径为0.5%三氟乙酸甲醇溶液和水的体积比为70∶30的流动相,柱规格为50
×
1000mm,紫外检测波长为205nm),以100ml/min的流速进行洗脱,收集洗脱液,每1000ml收集一瓶,分阶段收集gm1a和gm1b的洗脱液,并对洗脱液依据hplc高效液相色谱法(色谱条件:用十八烷基硅烷键合硅胶为填充剂,以0.01mol/l磷酸二氢钾溶液
‑
乙腈(32:68)为流动相a,以乙腈为流动相b;流速为每分钟1.2ml;检测波长为205nm)进行检测,将gm1a、gm1b洗脱液合并后减压浓缩至粘稠,加入浓缩液体积5倍量的丙酮,静置沉淀,静置沉淀,得到神经节苷脂粗品2;
81.(4)将粗品2加纯化水溶解后,加入5%(m/m)的针用活性炭,搅拌30min后,过滤,浓缩,加入丙酮沉淀,得神经节苷脂粗品3;
82.(3)用001
×
7强酸型阳离子树脂装柱(柱规格为50
×
300mm)后,使用盐酸水溶液(盐酸:纯化水=1l:10l)以70ml/min的流速冲洗阳离子交换柱3小时,将神经节苷脂粗品3溶解上样,使用盐酸水溶液进行洗脱,收集洗脱液,然后用10k中空纤维超滤膜超滤,至检测超滤液电导率≤10us/cm时停止超滤,得超滤液,将超滤液减压浓缩至粘稠后加入10倍量的丙酮,静置沉淀,将沉淀液过滤,干燥,得到本发明高纯度单唾液酸四己糖神经节苷脂钠。经测定,单唾液酸四己糖神经节苷脂钠的hplc纯度为99.5%。其所得产品的hplc谱图如图5所
示。
83.上述详细说明是针对本发明其中之一可行实施例的具体说明,该实施例并非用以限制本发明的专利范围,凡未脱离本发明所为的等效实施或变更,均应包含于本发明技术方案的范围内。