一种重组蛋白酶K的纯化方法及其应用与流程
本发明属于酶的分离提纯技术领域,更具体地,涉及一种重组蛋白酶k的纯化方法及其应用。
背景技术:
蛋白酶k最早于1974年在林伯式白色念球菌(tritirachiumalbumlimber)的提取物中发现,由于该酶能够消化富含二硫键的天然角蛋白(keratin),能够以角蛋白作为唯一的碳源和氮源生长,蛋白酶k也由此得名。
蛋白酶k是一种丝氨酸蛋白酶,其具有蛋白水解酶活性,对天然蛋白质有广谱的切割能力,偏好于切割脂肪族氨基酸和芳香族氨基酸,特别是丙氨酸的羧基端肽键。
蛋白酶k在一系列广泛的环境中均能保持活性,在ph4-12.5、高盐缓冲液及70℃的环境下也有活性,且能与变性剂和去污剂(如尿素、sds、盐酸胍、异硫氰酸胍、triton和tween)等兼容,且在一定浓度内,这些变性剂和去污剂能使蛋白酶k的活性得以提升。在医疗(病毒与微生物消杀)、食品(肉类嫩化)、皮革(毛发软化)、酿酒(酒类澄清)、氨基酸制备(降解羽毛)、核酸提取、原位杂交等方面,蛋白酶k均有应用。
随着蛋白酶k需求量的日益增大,其大规模生产显得尤为重要。具体地,蛋白酶k的大规模生产包括发酵、菌液分离、过滤、纯化、冻干和分装等过程。
早期,由于天然蛋白酶k表达量低,有一定的细胞毒性,获取足量的蛋白酶k粗酶液是限速步骤;伴随着突变体菌株的筛选、多拷贝数高表达蛋白酶k重组酵母菌株的筛选和发酵工艺的进一步优化,蛋白酶k发酵粗酶液的产能问题已逐步解决,从而导致其纯化是制约蛋白酶k规模化生产的关键。
蛋白酶k生产纯化工艺的报道不多,早期天然提取工艺中的纯化方法工艺复杂、纯化周期长且回收率低,不利于量产;重组表达蛋白酶k的开展后,采用在蛋白酶k的c末端添加6xhis标签,利于亲和纯化达到纯化的目的,该纯化方法的纯化介质成本较高,且存在ni脱落的风险,且实际工业生产中需进行三到四步才能取得理想的纯化效果。
综上所述,研究开发一种操作简单、成本低廉且回收率高的重组蛋白酶k的纯化方法,并将其应用到重组蛋白酶k的工业化分离提纯中,显得尤为重要,是本领域技术人员亟需解决的关键技术问题。
技术实现要素:
本发明的目的在于针对现有技术中存在的技术问题,提供一种重组蛋白酶k的纯化方法,在此基础上探讨其在重组蛋白酶k的工业化分离提纯中的应用,该纯化方法具有操作简单、成本低廉且回收率高的优势,实际应用效果优异。
为实现上述目的,本发明采用如下技术方案:
一种重组蛋白酶k的纯化方法,包括:
s1、往重组蛋白酶k发酵液中加入缓冲液a,混匀后一次超滤,获得已去除金属离子和分子量小于20kda杂蛋白的超滤液;
s2、将步骤s1中得到的超滤液泵入用缓冲液a平衡的阳离子交换柱中,控制流速为0.35-0.6l/min,随后用缓冲液a复平衡后用缓冲液b洗脱,收集洗脱液;
s3、往洗脱液中加入缓冲液c,混匀后二次超滤,即可。
其中,上述缓冲液a、缓冲液b和缓冲液c分别为:
缓冲液a:35-60mmmes,30-55mmnacl,1.5-3mmedta-2na,ph5.5-6.5;
缓冲液b:35-60mmmes,135-170mmnacl,ph5.5-6.5;
缓冲液c:15-25mmtris-hcl,20-40mmnacl,1.5-5mmcacl2,ph6.0-6.8。
详细地,在步骤s1的一次超滤中,因为发酵液中离子强度高,经过缓冲液a换液,降低上样前蛋白的离子浓度;蛋白酶k的等电点在ph8.9左右,在ph5.6-6.5时,蛋白带正电荷,所以在低盐离子缓冲液时可以结合到阳离子交换介质上。在步骤s2中,用高离子强度的缓冲液b将蛋白洗脱下来。进一步地,为了达到工业需求,还需要进行步骤s3的二次超滤,换到常规的缓冲液c中,该缓冲液有利于蛋白酶k的后续应用。
具体地,在缓冲液a中添加edta,可以除去在核酸提取时影响核酸质量的金属离子;缓冲液c的ph如果调整在7.4左右,步骤s3在换液过程中就很容易出现沉淀,导致换液失败;而缓冲液c在ph6.0-6.8时换液比较稳定,其原因在于,缓冲液c的ph值与蛋白酶k等电点相差较远。
在上述技术方案中,步骤s1中,所述加入缓冲液a后一次超滤的次数为2-6次,优选为4次。
进一步地,在上述技术方案中,步骤s1中,所述缓冲液a的加入体积为所述重组蛋白酶k发酵液的0.8-2倍,优选为1-1.2倍。
详细地,在步骤s1的一次超滤中,缓冲液a的换液总体积大于等于原始体积的16倍即可。如果换液总体积不够,离子强度还没有降(到cond6.0±0.5ms/cm)下来,会影响蛋白酶k与离子交换介质的结合效率;如果换液总体积到原始体积的32倍及以上,会增加了该步骤的操作时间,并且也不会进一步改善产品质量,从时间成本上看不划算,并且,随着换液次数增加,蛋白酶k也会损失,回收率会下降。
在本发明的一个优选实施方式中,步骤s1中,还包括,在加入缓冲液a混匀和一次超滤前,用亲水性聚醚砜囊式滤芯对所述重组蛋白酶k发酵液进行过滤。
优选地,在上述技术方案中,所述亲水性聚醚砜囊式滤芯的孔径为0.22μm。
在上述技术方案中,步骤s3中,所述缓冲液c的加入体积为所述洗脱液的2.5-4倍,优选为3-3.5倍。
进一步地,在上述技术方案中,步骤s3中,所述加入缓冲液c后二次超滤的次数为2-4次,优选为3次。
再进一步地,在上述技术方案中,步骤s2中,所述阳离子交换柱的阳离子交换介质为spbestaroseff、unosphererapids和porostmxs中的一种。
又进一步地,在上述技术方案中,所述一次超滤和/或所述二次超滤为膜包超滤和中空纤维超滤膜超滤中的一种。
在本发明的一个具体实施方式中,所述重组蛋白酶k的纯化方法具体包括以下步骤:
(1)将重组蛋白酶k发酵液经板框过滤后收集并置于2-8℃冷藏,纯化前,用0.22um的亲水性聚醚砜囊式滤芯过滤进一步除去细胞碎片及其他杂质,将滤液与缓冲液a按体积比为1:1混匀后,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,待体积浓缩到初始体积的一半时,再次加入相同体积的缓冲液a混匀,再用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,连续重复4次,除去其中的金属离子和分子量小于20kda的杂蛋白;
(2)以利穗appspilot全自动蛋白纯化仪进行阳离子交换纯化(上海博格隆spbestaroseff纯化介质),具体为,用超纯水清洗好系统后,用缓冲液a平衡阳离子交换柱,平衡结束后用流速0.5l/min流速上样,上样结束后用缓冲液a复平衡,最后用缓冲液b洗脱,收集洗脱液;
(3)用3倍体积的缓冲液c稀释步骤(2)中得到的洗脱液,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,待体积浓缩到初始体积的四分之一时,再次加入相同体积的缓冲液c混匀,再用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,连续重复3次,即得纯化后的重组蛋白酶k。
其中,上述缓冲液a、缓冲液b和缓冲液c分别为:
缓冲液a:50mmmes,40mmnacl,2mmedta-2na,ph5.9-6.1;
缓冲液b:50mmmes,150mmnacl,ph5.9-6.1;
缓冲液c:20mmtris-hcl,30mmnacl,2mmcacl2,ph6.2-6.4。
本发明又一方面还提供了上述重组蛋白酶k的纯化方法在重组蛋白酶k的分离和提纯中的应用。
与现有技术相比,本发明的有益效果为:
(1)本发明所提供的重组蛋白酶k的纯化方法仅采用简单的超滤-离子交换-超滤的纯化工艺就满足产品需求,非常适合工业级生产,当采用苏州利穗appspilot全自动蛋白纯化仪,每日可达到500g左右的生产规模;
(2)本发明所提供的重组蛋白酶k的纯化方法所用试剂的原料成本低廉且蛋白回收率高,回收率达69%左右;
(3)本发明所提供的重组蛋白酶k的纯化方法制备得到的蛋白酶k的sds-page纯度在95%以上,酶活性在800u/ml以上,比活性在40u/mg以上。
附图说明
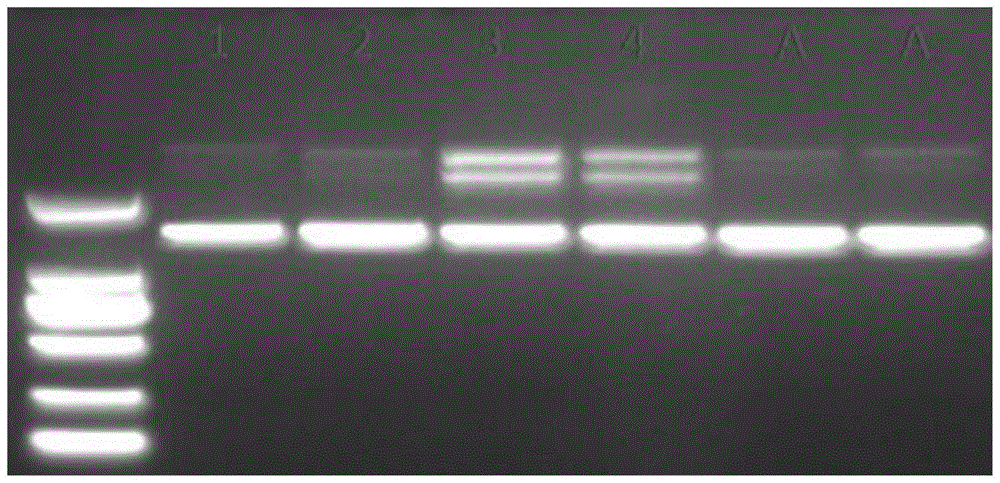
图1所示为本发明实施例的金属离子残留检测结果图;
图2所示为本发明实施例的蛋白酶k的dna酶残留检测结果的电泳图;
图3所示为本发明实施例的蛋白酶k的rna酶残留检测结果的电泳图。
具体实施方式
下面结合具体实施例对本发明作进一步的详细说明,以使本领域的技术人员更加清楚地理解本发明。
以下实施例,仅用于说明本发明,但不止用来限制本发明的范围。
基于本发明中的具体实施例,本领域普通技术人员在没有做出创造性劳动的情况下,所获得的其他所有实施例,都属于本发明的保护范围。
在本发明实施例中,若无特殊说明,所有原料组分均为本领域技术人员熟知的市售产品。
在本发明实施例中,若无特殊说明,所用技术手段均为本领域技术人员熟知的常规技术手段。
在本发明实施例中,所用的原料和相关设备信息如下表所示。
实施例1
本发明实施例提供了一种重组蛋白酶k的纯化方法,具体包括以下步骤:
(1)将重组蛋白酶k发酵液经板框过滤后收集并置于2-8℃冷藏,纯化前,用0.22um的亲水性聚醚砜囊式滤芯过滤进一步除去细胞碎片及其他杂质,将滤液与缓冲液a按体积比为1:1混匀后,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,待体积浓缩到初始体积的一半时,再次加入相同体积的缓冲液a混匀,再用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,连续重复4次,除去其中的金属离子和分子量小于20kda的杂蛋白;
(2)以利穗appspilot全自动蛋白纯化仪进行阳离子交换纯化(上海博格隆spbestaroseff纯化介质),具体为,用超纯水清洗好系统后,用缓冲液a平衡阳离子交换柱,平衡结束后用流速0.5l/min流速上样,上样结束后用缓冲液a复平衡,最后用缓冲液b洗脱,收集洗脱液;
(3)用3倍体积的缓冲液c稀释步骤(2)中得到的洗脱液,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,待体积浓缩到初始体积的四分之一时,再次加入相同体积的缓冲液c混匀,再用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,连续重复3次,即得纯化后的重组蛋白酶k。
其中,上述缓冲液a、缓冲液b和缓冲液c分别为:
缓冲液a:50mmmes,40mmnacl,2mmedta-2na,ph5.9-6.1;
缓冲液b:50mmmes,150mmnacl,ph5.9-6.1;
缓冲液c:20mmtris-hcl,30mmnacl,2mmcacl2,ph6.2-6.4。
对比例1
本发明对比例提供了一种重组蛋白酶k的纯化方法,与实施例1相比,其区别仅在于缓冲液a1中不含有edta-2na,具体包括以下步骤:
(1)将重组蛋白酶k发酵液经板框过滤后收集并置于2-8℃冷藏,纯化前,用0.22um的亲水性聚醚砜囊式滤芯过滤进一步除去细胞碎片及其他杂质,将滤液与缓冲液a1按体积比为1:1混匀后,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,待体积浓缩到初始体积的一半时,再次加入相同体积的缓冲液a1混匀,再用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,连续重复4次,除去其中的金属离子和分子量小于20kda的杂蛋白;
(2)以利穗appspilot全自动蛋白纯化仪进行阳离子交换纯化(上海博格隆spbestaroseff纯化介质),具体为,用超纯水清洗好系统后,用缓冲液a1平衡阳离子交换柱,平衡结束后用流速0.5l/min流速上样,上样结束后用缓冲液a1复平衡,最后用缓冲液b洗脱,收集洗脱液;
(3)用3倍体积的缓冲液c稀释步骤(2)中得到的洗脱液,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,待体积浓缩到初始体积的四分之一时,再次加入相同体积的缓冲液c混匀,再用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,连续重复3次,即得纯化后的重组蛋白酶k。
其中,上述缓冲液a1、缓冲液b和缓冲液c分别为:
缓冲液a1:50mmmes,40mmnacl,ph5.9-6.1;
缓冲液b:50mmmes,150mmnacl,ph5.9-6.1;
缓冲液c:20mmtris-hcl,30mmnacl,2mmcacl2,ph6.2-6.4。
对比例2
本发明对比例提供了一种重组蛋白酶k的纯化方法,与实施例1相比,其区别仅在于缓冲液c1的ph调整为7.3-7.5,具体包括以下步骤:
(1)将重组蛋白酶k发酵液经板框过滤后收集并置于2-8℃冷藏,纯化前,用0.22um的亲水性聚醚砜囊式滤芯过滤进一步除去细胞碎片及其他杂质,将滤液与缓冲液a按体积比为1:1混匀后,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,待体积浓缩到初始体积的一半时,再次加入相同体积的缓冲液a混匀,再用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,连续重复4次,除去其中的金属离子和分子量小于20kda的杂蛋白;
(2)以利穗appspilot全自动蛋白纯化仪进行阳离子交换纯化(上海博格隆spbestaroseff纯化介质),具体为,用超纯水清洗好系统后,用缓冲液a平衡阳离子交换柱,平衡结束后用流速0.5l/min流速上样,上样结束后用缓冲液a复平衡,最后用缓冲液b洗脱,收集洗脱液;
(3)用3倍体积的缓冲液c1稀释步骤(2)中得到的洗脱液,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,在浓缩过程中,一般还未到初始体积一半时即出现沉淀,换液步骤以失败结束。
其中,上述缓冲液a、缓冲液b和缓冲液c1分别为:
缓冲液a:50mmmes,40mmnacl,2mmedta-2na,ph5.9-6.1;
缓冲液b:50mmmes,150mmnacl,ph5.9-6.1;
缓冲液c:20mmtris-hcl,30mmnacl,2mmcacl2,ph7.3-7.5。
对比例3
本发明对比例提供了一种重组蛋白酶k的纯化方法,与实施例1相比,其区别仅在于步骤s1中的一次超滤得次数为7次,具体包括以下步骤:
(1)将重组蛋白酶k发酵液经板框过滤后收集并置于-4℃下冷藏,纯化前,用0.22um的亲水性聚醚砜囊式滤芯过滤进一步除去细胞碎片及其他杂质,将滤液与缓冲液a1按体积比为1:1混匀后,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,待体积浓缩到初始体积的一半时,再次加入相同体积的缓冲液a1混匀,再用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,连续重复7次,除去其中的金属离子和分子量低于约20kd的杂蛋白;
(2)以利穗appspilot全自动蛋白纯化仪进行阳离子交换纯化(上海博格隆spbestaroseff纯化介质),具体为,用超纯水清洗好系统后,用缓冲液a1平衡阳离子交换柱,平衡结束后用流速0.5l/min流速上样,上样结束后用缓冲液a1复平衡,最后用缓冲液b洗脱,收集洗脱液;
(3)用3倍体积的缓冲液c稀释步骤(2)中得到的洗脱液,用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,待体积浓缩到初始体积的四分之一时,再次加入相同体积的缓冲液c混匀,再用截留8kda、表面积2.5m2的超滤膜包换液和浓缩,连续重复3次,即得纯化后的重组蛋白酶k。
其中,上述缓冲液a1、缓冲液b和缓冲液c分别为:
缓冲液a:50mmmes,40mmnacl,2mmedta-2na,ph5.9-6.1;
缓冲液b:50mmmes,150mmnacl,ph5.9-6.1;
缓冲液c:20mmtris-hcl,30mmnacl,2mmcacl2,ph6.2-6.4。
试验结果
(1)回收率计算
根据体积和总蛋白的浓度,计算出总蛋白的量,然后,与原始初酶液(回收率按照100%计算)对比,计算出每一步骤后的回收率。
具体结果如下表所示:
分析上表可见,对比例3中增加了超滤换液的次数,会影响最终产品的回收率,还增加了该步骤的时间;同时,可以看出本发明实施例的蛋白酶k的回收率在69%左右。
(2)活性测定
分别采用实施例1和对比例1的方法对武汉爱博泰克生物科技有限公司的产品进行纯化后,与市售产品1(江苏金普诺安蛋白酶k产品)和市售产品2(sigma蛋白酶k产品)分别配制成20mg/ml的溶液后,测定酶活性和比活性测定。
活性测定方法参考酪蛋白为底物的,folin&ciocalteu'sphenol试剂的分光光度计法(具体可参考sigma官网)。
测试结果如下表所示:
从活性测定,可见本发明实施例纯化出的蛋白酶k酶活性,比活性与市售产品1相当,略高于市售产品2。
(3)金属离子残留检测
分别用实施例1和对比例1的方法获取的蛋白酶k产品,市售产品1(江苏金普诺安蛋白酶k产品)与质粒puc19孵育,观察dna是否稳定。即:将蛋白酶k溶液与无dna酶污染的puc19质粒37℃孵育16小时,琼脂糖凝胶电泳检测质粒是否完整,20ul反应体系如下:单位(ul)
检测结果如图1所示。图中,1、2泳道对应于采用实施例1的方法对武汉爱博泰克生物科技有限公司的产品进行纯化后的样品,3、4泳道对应于采用对比例1的方法对武汉爱博泰克生物科技有限公司的产品进行纯化后的样品,a泳道对应于市售产品1(江苏金普诺安蛋白酶k产品);从图中可以看出,如果缓冲液a1中不添加edta-2na,可能造成金属离子超标,导致dna非特异性断裂。
(4)蛋白酶k的dna酶残留检测
分别将采用实施例1的方法对武汉爱博泰克生物科技有限公司的产品进行纯化后得到的样品,与市售产品1(江苏金普诺安蛋白酶k产品)和市售产品2(sigma蛋白酶k产品)的蛋白酶k溶液与无dna酶污染的puc19质粒37℃孵育16小时,检测是否有dna酶残留,同时设置空白对照组,20ul反应体系如下:单位(ul)
待反应结束后,添加6x的dna上样buffer终止反应,用0.7%的琼脂糖凝胶电泳。
电泳图如图2所示;图中,1、2泳道对应于市售产品1(江苏金普诺安蛋白酶k产品);3、4泳道对应于市售产品2(sigma蛋白酶k产品);5、6泳道对应于采用实施例1的方法对武汉爱博泰克生物科技有限公司的产品进行纯化后的样品;7、8泳道对应于空白对照组;m是dnamarker。从图中可以看出,采用实施例1的方法对武汉爱博泰克生物科技有限公司的产品进行纯化后得到的蛋白酶k样品无dna酶残留污染。
(5)蛋白酶k的rna酶残留检测
分别将采用实施例1的方法对武汉爱博泰克生物科技有限公司的产品进行纯化后得到的样品,与市售产品1(江苏金普诺安蛋白酶k产品)和市售产品2(sigma蛋白酶k产品)的蛋白酶k溶液与无rna酶污染的最新提取的rna于37℃孵育16小时,检测是否有rna酶残留,同时设置空白对照组,20ul反应体系如下:单位(ul)
待反应结束后,添加6x的rna上样buffer终止反应,用0.7%的琼脂糖凝胶电泳。
电泳图如图3所示;图中,1、2泳道对应于市售产品1(江苏金普诺安蛋白酶k产品);3、4泳道对应于市售产品2(sigma蛋白酶k产品);5、6泳道对应于采用实施例1的方法对武汉爱博泰克生物科技有限公司的产品进行纯化后的样品;7、8泳道对应于空白对照组;9是dnamarker。从图中可以看出,采用实施例1的方法对武汉爱博泰克生物科技有限公司的产品进行纯化后得到的蛋白酶k样品无rna酶残留污染。
最后,本发明的方法仅为较佳的实施方案,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!