苯基嘧啶胺类抗肿瘤化合物及其制备方法和应用
1.本法发明属于医药技术领域,涉及一类具有抗肿瘤活性的特定化学结构的化合物,具体涉及苯基嘧啶胺类抗肿瘤化合物及其制备方法和应用。
背景技术:
2.恶性黑色素瘤是侵袭能力最强,转移能力最高的皮肤癌症,实验证明,恶性黑色素瘤的自然凋亡率明显低于其他肿瘤。虽然黑色素瘤仅占有皮肤癌的5%,但80%的皮肤癌死亡病例与黑色素瘤相关。
3.实验证明,内皮素b受体在恶性黑色素瘤细胞中表达增加显著,当内皮素1、内皮素3与内皮素b受体结合后,产生多种有助于增殖和侵袭的生物学效应,如e
‑
钙黏蛋白和相关连接蛋白表达下调,n
‑
钙黏蛋白表达上调,缝隙连接蛋白cx43磷酸化,α
v
β3整合素、α2β1整合素、基质金属蛋白酶2基质、金属蛋白酶9表达增加,以及下游的粘着斑激酶通路和细胞外信号调节激酶1/2信号通路激活。而且这些生物学效应均可被内皮素受体b拮抗剂显著抑制,从而抑制了恶性肿瘤的增殖和转移能力。而且恶性黑色素瘤细胞的表型和基因型分析鉴定出内皮素b受体为肿瘤标志物,使其成为潜在的治疗恶性黑色素瘤的靶点。
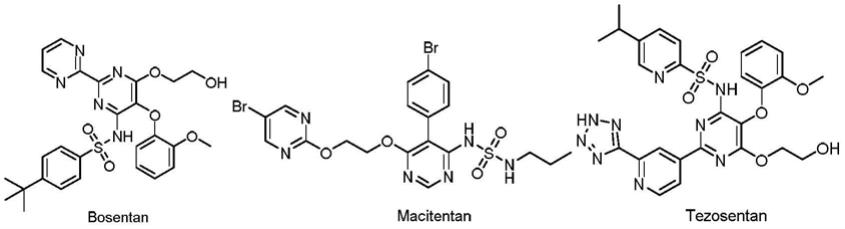
4.目前有很多非肽类内皮素受体拮抗剂,但它们主要用于高血压的治疗。如用于治疗who功能分级ii级
‑
iv级的肺动脉高压的波生坦,用于治疗肺动脉高血压的马西替坦,以及研究用于充血性心力衰竭,肝病和心脏病的使用和治疗的替唑生坦。
[0005][0006]
根据上述的内皮素b受体拮抗剂的公共母核进行优化,将与嘧啶环相连的部分直接提取出来,以期得到抗肿瘤活性更好的全新化合物,现有技术并未见到相关结构的报道。
技术实现要素:
[0007]
鉴于现有技术存在的问题,本发明的目的在于提供一种苯基嘧啶胺类化合物及其制备方法和应用,所制备的化合物在体外抗肿瘤活性测试中显现出了良好的结果,可用于制备抗肿瘤药物。
[0008]
为实现上述目的,本发明采用以下技术方案。
[0009]
一种苯基嘧啶胺类化合物,所述化合物的结构通式i如下:
[0010][0011]
通式i
[0012]
其中:r基团为氢原子、2位单取代的氟原子,或3位、4位单取代的甲基、氟原子、氯原子、溴原子。
[0013]
进一步地,所述的苯基嘧啶胺类化合物,所述通式为ⅰ的化合物或其药学上可接受的盐、水合物或溶剂化物,结构选自下述任意一种:
[0014]6‑
氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
苯基嘧啶
‑4‑
胺(a1);
[0015]6‑
氯
‑2‑
(2
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a2);
[0016]6‑
氯
‑2‑
(3
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a3);
[0017]6‑
氯
‑2‑
(4
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a4);
[0018]6‑
氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(间甲苯基)嘧啶
‑4‑
胺(a5);
[0019]6‑
氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(对甲苯基)嘧啶
‑4‑
胺(a6);
[0020]6‑
氯
‑2‑
(3
‑
氯苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a7);
[0021]6‑
氯
‑2‑
(4
‑
氯苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a8);
[0022]2‑
(3
‑
溴苯基)
‑6‑
氯
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a9);
[0023]2‑
(4
‑
溴苯基)
‑6‑
氯
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a10)。
[0024]
但不仅限于以上化合物,只要化合物结构式满足通式,均为本发明的限定范围。
[0025]
所述苯基嘧啶胺类化合物的制备方法,具体包括以下步骤。
[0026]
步骤1、将1倍量丙二酸二乙酯、1.05倍量nbs和适量三氯甲烷放入反应瓶中,再加入微量浓硫酸作为催化剂,在50℃的条件下反应10
‑
12h;薄层色谱监控反应进程,反应完毕后,用饱和的亚硫酸钠溶液多次洗涤反应液,再用饱和的氯化钠溶液多次洗涤反应液,用无水硫酸钠干燥反应液,然后减压蒸出溶剂,得2
‑
溴丙二酸二乙酯,为无色透明液体。
[0027]
步骤2、将1倍量2
‑
溴丙二酸二乙酯、1倍量愈创木酚,1.5倍量碳酸钾和适量乙腈放入反应瓶中,在80℃的条件下反应10
‑
12h,薄层色谱监控反应进程;反应完毕后,将反应液抽滤,滤饼用乙腈溶液洗涤,再次抽滤,合并所得滤液,减压蒸出溶剂,得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,为黑色液体。
[0028]
步骤3、将1倍量r取代的苯甲腈、0.6倍量甲醇钠和适量甲醇放入反应瓶中,室温反应16h;加入1.8倍量氯化铵,室温继续反应10
‑
12h,薄层色谱监控反应进程;反应完毕后,减压蒸出溶剂,得白色固体;用适量乙醇溶剂溶解固体,超声,抽滤,收集滤液,减压蒸出溶剂,得r取代的苯甲脒盐酸盐,为白色固体。
[0029]
步骤4、将1.5倍量金属钠切成条状,在低温下缓慢加入盛有适量甲醇的反应瓶中,
反应0.5h,然后加入1.3倍量2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯和1倍量r取代的苯甲脒盐酸盐,室温反应18小时,薄层色谱监控反应进程,反应完毕后,减压蒸出溶剂,然后加入氢氧化钠溶液,抽滤,滤液使用盐酸调ph值至酸性,析出大量固体,抽滤,滤饼干燥,得5
‑
(2
‑
甲氧基苯氧基)
‑2‑
(r取代苯基)嘧啶
‑
4,6
‑
二醇,为淡黄色固体。
[0030]
步骤5、将5
‑
(2
‑
甲氧基苯氧基)
‑2‑
(r取代苯基)嘧啶
‑
4,6
‑
二醇和适量三氯氧磷加入反应瓶中,在100℃下反应48小时;反应完毕后,将反应液缓慢倒入冰水中,析出固体,抽滤,滤饼干燥,得4,6
‑
二氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(r取代苯基)嘧啶,为黑色固体。
[0031]
步骤6、将1倍量4,6
‑
二氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(r取代苯基)嘧啶、1倍量氯化铵、2倍量碳酸钾和适量dmf放入反应瓶中,80℃下反应10
‑
12h,薄层色谱监控反应进程;反应完毕后,将反应液缓慢倒入水中,析出固体,抽滤,滤饼干燥,使用柱色谱法除杂,然后使用异丙醇/水重结晶,得6
‑
氯
‑2‑
(r取代苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺,为白色固体。
[0032]
一种药物组合物包括所述的苯基嘧啶胺类化合物、其药学上可接受的盐、水合物或溶剂化物和药学上可接受的载体。
[0033]
所述的苯基嘧啶胺类化合物或其药学上可接受的盐、水合物或溶剂化物或所述的药物组合物在制备治疗抗肿瘤药物中的应用。
[0034]
进一步地,所述的肿瘤为恶性黑色素瘤。
[0035]
进一步地,所述药物的剂型为药物治疗学上可接受的剂型。
[0036]
进一步地,所述药物的剂量为药物治疗学上可接受的剂量。
[0037]
本发明还包括本发明化合物的前药。本发明化合物的前药是通式i的衍生物,在生理条件下(例如通过代谢、溶剂分解或另外的方式)被转化成相应的生物活性形式。本发明的药用组合物可配制成若干种剂型,所述剂型包括但不限于注射剂、片剂、胶囊剂、散剂等。
[0038]
与现有技术比,本发明的有益效果如下。
[0039]
本发明提供的苯基嘧啶胺类化合物在体外抗肿瘤活性测试中显现出了良好的结果,对人恶性黑色素瘤细胞有一定的抑制活性,可用于制备抗肿瘤药物,为开发以内皮素受体为新靶点的抗肿瘤药物开辟了新的途径。本发明提供制备方法简单可行,收率较高,易于大规模生产。
具体实施方式
[0040]
下面结合具体实施例对本发明作进一步的说明,这些实施例只是举例说明,绝不限制本发明的范围。
[0041]
一种苯基嘧啶胺类化合物,所述化合物的结构通式i如下:
[0042][0043]
通式i
[0044]
其中:r基团为氢原子、2位单取代的氟原子,或3位、4位单取代的甲基、氟原子、氯原子、溴原子。
[0045]
实施例1 6
‑
氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
苯基嘧啶
‑4‑
胺(a1)的制备。
[0046]
a.2
‑
溴丙二酸二乙酯的制备。
[0047]
将丙二酸二乙酯(30.00g,187.30mmol),nbs(35.00g,196.67mmol)和100ml三氯甲烷放入250ml烧瓶中,再加入两滴浓硫酸作为催化剂。在50℃的条件下反应12h,薄层色谱监控反应进程。反应完毕后,用饱和的亚硫酸钠溶液洗涤反应液,再用饱和的氯化钠溶液洗涤反应液,然后用无水硫酸钠干燥反应液,减压蒸出溶剂,得无色透明液体产物43.65g,收率97.48%。
[0048]
b.2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯的制备。
[0049]
将2
‑
溴丙二酸二乙酯(43.65g,182.59mmol),愈创木酚(22.67g,182.59mmol),碳酸钾(35.33g,255.62mmol)和100ml乙腈放入250ml烧瓶中,在80℃的条件下反应13h,薄层色谱监控反应进程。反应完毕后,将反应液抽滤,滤饼用乙腈洗涤,再次抽滤,合并滤液,减压蒸出溶剂,得黑色液体产物45.27g,收率87.83%。
[0050]
c.苯甲脒盐酸盐的制备。
[0051]
将苯甲腈(10.00g,96.97mmol),甲醇钠(3.14g,58.18mmol)和100ml甲醇放入250ml烧瓶中,室温反应16h,再加入氯化铵(9.34g,174.55mmol),室温继续反应12h,薄层色谱监控反应进程。反应完毕后,减压蒸出溶剂,然后用乙醇分散固体,超声,抽滤,收集滤液,减压蒸出溶剂,得白色固体产物15.02g,收率98.88%。
[0052]
d.5
‑
(2
‑
甲氧基苯氧基)
‑2‑
苯基嘧啶
‑
4,6
‑
二醇的制备。
[0053]
将固体钠(3.31g,143.86mmol)切成条状,在冷阱中缓慢加入盛有100ml甲醇的250ml烧瓶中,反应0.5h,然后加入2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯(35.20g,124.68mmol)和苯甲脒盐酸盐(15.02g,95.91mmol),室温反应18h,薄层色谱监控反应进程。反应完毕后,减压蒸出溶剂,然后加入ph值为9.0的氢氧化钠溶液溶解固体,抽滤,滤液使用稀盐酸调ph值至3.0,析出大量固体,抽滤,滤饼干燥,得淡黄色固体14.88g,收率50.00%。
[0054]
e.4,6
‑
二氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
苯基嘧啶的制备。
[0055]
将5
‑
(2
‑
甲氧基苯氧基)
‑2‑
苯基嘧啶
‑
4,6
‑
二醇(14.88g,47.95mmol)和50ml三氯氧磷加入250ml烧瓶中,在100℃下反应48h。反应完毕后,将反应液缓慢倒入冰水中,析出黑色固体,抽滤,滤饼干燥,得黑色固体15.21g,收率91.35%。
[0056]
f.6
‑
氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
苯基嘧啶
‑4‑
胺的制备。
[0057]
将4,6
‑
二氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
苯基嘧啶(15.21g,43.81mmol),氯化铵(2.34g,43.81mmol),碳酸钾(12.11g,87.62mmol)和100ml dmf放入250ml烧瓶中,80℃下反应12h,薄层色谱监控反应进程。反应完毕后,将反应液倒入水中,析出固体,抽滤,滤饼干燥。使用柱色谱法提纯,然后使用异丙醇/水重结晶,得白色固体产品3.26g,收率23.78%。
[0058]1h nmr(500mhz,dmso
‑
d6)δ8.28
–
8.23(m,2h),7.50(d,j=4.9hz,3h),7.34(s,2h),7.12(d,j=8.0hz,1h),7.04(t,j=7.7hz,1h),6.84(t,j=7.7hz,1h),6.65(d,j=8.1hz,1h),3.86(s,3h)。
[0059]
实施例2 6
‑
氯
‑2‑
(2
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a2)的制备。
[0060]
以丙二酸二乙酯为原料,按照实施例1a,1b步骤制得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,再以2
‑
氟苯腈为原料,按照实施例1c,1d,1e步骤制得4,6
‑
二氯
‑2‑
(2
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶,按照实施例1f步骤制得6
‑
氯
‑2‑
(2
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a2),白色固体,收率:22.17%。
[0061]1h nmr(500mhz,dmso
‑
d6)δ7.91(t,j=7.8hz,1h),7.57
–
7.20(m,5h),7.13(d,j=8.1hz,1h),7.05(t,j=7.7hz,1h),6.86(t,j=7.7hz,1h),6.65(d,j=8.0hz,1h),3.86(s,3h)。
[0062]
实施例3 6
‑
氯
‑2‑
(3
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a3)的制备。
[0063]
以丙二酸二乙酯为原料,按照实施例1a,1b步骤制得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,再以3
‑
氟苯腈为原料,按照实施例1c,1d,1e步骤制得4,6
‑
二氯
‑2‑
(3
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶,按照实施例1f步骤制得6
‑
氯
‑2‑
(3
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a3),淡黄色固体,收率:23.52%。
[0064]1h nmr(500mhz,dmso
‑
d6)δ8.09(d,j=7.8hz,1h),7.95(d,j=9.5hz,1h),7.71
–
7.23(m,4h),7.12(d,j=8.1hz,1h),7.04(t,j=7.7hz,1h),6.84(t,j=7.6hz,1h),6.66(d,j=8.0hz,1h),3.86(s,3h)。
[0065]
实施例4 6
‑
氯
‑2‑
(4
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a4)的制备。
[0066]
以丙二酸二乙酯为原料,按照实施例1a,1b步骤制得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,再以4
‑
氟苯腈为原料,按照实施例1c,1d,1e步骤制得4,6
‑
二氯
‑2‑
(4
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶,按照实施例1f步骤制得6
‑
氯
‑2‑
(4
‑
氟苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a4),淡黄色固体,收率:24.17%。
[0067]1h nmr(500mhz,dmso
‑
d6)δ8.28(t,2h),7.38(s,2h),7.32(t,j=8.3hz,2h),7.12(d,j=8.1hz,1h),7.04(t,j=7.7hz,1h),6.83(t,j=7.7hz,1h),6.65(d,j=8.0hz,1h),3.86(s,3h)。
[0068]
实施例5 6
‑
氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(间甲苯基)嘧啶
‑4‑
胺(a5)的制备。
[0069]
以丙二酸二乙酯为原料,按照实施例1a,1b步骤制得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,再以3
‑
甲基苯甲腈为原料,按照实施例1c,1d,1e步骤制得4,6
‑
二氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(间甲苯基)嘧啶,按照实施例1f步骤制得6
‑
氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(间甲苯基)嘧啶
‑4‑
胺(a5),淡黄色固体,收率23.28%。
[0070]1h nmr(500mhz,dmso
‑
d6)δ8.08(s,1h),8.04(d,j=7.6hz,1h),7.53
–
7.20(m,4h),7.12(d,j=8.1hz,1h),7.04(t,j=7.7hz,1h),6.84(t,j=7.7hz,1h),6.64(d,j=8.0hz,
1h),3.86(s,3h),2.39(s,3h)。
[0071]
实施例6 6
‑
氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(对甲苯基)嘧啶
‑4‑
胺(a6)的制备。
[0072]
以丙二酸二乙酯为原料,按照实施例1a,1b步骤制得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,再以4
‑
甲基苯甲腈为原料,按照实施例1c,1d,1e步骤制得4,6
‑
二氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(对甲苯基)嘧啶,按照实施例1f步骤制得6
‑
氯
‑5‑
(2
‑
甲氧基苯氧基)
‑2‑
(对甲苯基)嘧啶
‑4‑
胺(a6),白色固体,收率24.43%。
[0073]1h nmr(500mhz,dmso
‑
d6)δ8.15(d,j=7.7hz,2h),7.49
–
7.19(m,4h),7.12(d,j=8.1hz,1h),7.03(t,j=7.7hz,1h),6.83(t,j=7.7hz,1h),6.64(d,j=8.0hz,1h),3.86(s,3h),2.37(s,3h)。
[0074]
实施例7 6
‑
氯
‑2‑
(3
‑
氯苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a7)的制备。
[0075]
以丙二酸二乙酯为原料,按照实施例1a,1b步骤制得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,再以3
‑
氯苯甲腈为原料,按照实施例1c,1d,1e步骤制得4,6
‑
二氯
‑2‑
(3
‑
氯苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶,按照实施例1f步骤制得6
‑
氯
‑2‑
(3
‑
氯苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a7),淡黄色固体,收率25.83%。
[0076]1h nmr(500mhz,dmso
‑
d6)δ8.23(s,1h),8.18(d,j=7.6hz,1h),7.76
–
7.18(m,4h),7.12(d,j=7.9hz,1h),7.04(t,j=7.7hz,1h),6.83(t,j=7.6hz,1h),6.66(d,j=8.0hz,1h),3.86(s,3h)。
[0077]
实施例8 6
‑
氯
‑2‑
(4
‑
氯苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a8)的制备。
[0078]
以丙二酸二乙酯为原料,按照实施例1a,1b步骤制得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,再以4
‑
氯苯甲腈为原料,按照实施例1c,1d,1e步骤制得4,6
‑
二氯
‑2‑
(4
‑
氯苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶,按照实施例1f步骤制得6
‑
氯
‑2‑
(4
‑
氯苯基)
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a8),淡黄色固体,收率25.16%。
[0079]1h nmr(500mhz,dmso
‑
d6)δ8.25(d,j=8.1hz,2h),7.56(d,j=8.1hz,2h),7.40(s,2h),7.12(d,j=8.0hz,1h),7.04(t,j=7.7hz,1h),6.83(t,j=7.7hz,1h),6.66(d,j=8.0hz,1h),3.86(s,3h)。
[0080]
实施例9 2
‑
(3
‑
溴苯基)
‑6‑
氯
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a9)的制备。
[0081]
以丙二酸二乙酯为原料,按照实施例1a,1b步骤制得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,再以3
‑
溴苯腈为原料,按照实施例1c,1d,1e步骤制得2
‑
(3
‑
溴苯基)
‑
4,6
‑
二氯
‑5‑
(2
‑
甲氧基苯氧基)嘧啶,按照实施例1f步骤制得2
‑
(3
‑
溴苯基)
‑6‑
氯
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a9),白色固体,收率28.86%。
[0082]1h nmr(500mhz,dmso
‑
d6)δ8.39(s,1h),8.22(d,j=7.7hz,1h),7.82
–
7.18(m,4h),7.12(d,j=8.1hz,1h),7.04(t,j=7.7hz,1h),6.83(t,j=7.7hz,1h),6.66(d,j=8.0hz,1h),3.86(s,3h)。
[0083]
实施例10 2
‑
(4
‑
溴苯基)
‑6‑
氯
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a10)的制备。
[0084]
以丙二酸二乙酯为原料,按照实施例1a,1b步骤制得2
‑
(2
‑
甲氧基苯氧基)丙二酸二乙酯,再以4
‑
溴苯腈为原料,按照实施例1c,1d,1e步骤制得2
‑
(4
‑
溴苯基)
‑
4,6
‑
二氯
‑5‑
(2
‑
甲氧基苯氧基)嘧啶,按照实施例1f步骤制得2
‑
(4
‑
溴苯基)
‑6‑
氯
‑5‑
(2
‑
甲氧基苯氧基)嘧啶
‑4‑
胺(a10),白色固体,收率27.61%。
[0085]1h nmr(500mhz,dmso
‑
d6)δ8.17(d,j=7.0hz,2h),7.70(d,j=7.0hz,2h),7.43(s,
2h),7.12(d,j=8.0hz,1h),7.04(t,j=7.1hz,1h),6.82(t,j=7.5hz,1h),6.65(d,j=8.0hz,1h),3.85(s,3h)。
[0086]
实施例11抑制肿瘤细胞增殖实验。
[0087]
对本发明的化合物进行了肿瘤细胞增殖抑制实验,试验方法采用常规的mtt法。
[0088]
肿瘤细胞的培养:细胞株选用sk
‑
mel
‑
28(人皮肤恶性黑色素瘤细胞)、a375(人黑色素瘤细胞),以dmem+10%fbs+双抗(青霉素100单位/ml,链霉素100μg/ml)的培养液培养。
[0089]
样品配制:用dmso(merck)溶解后,加入pbs(
‑
)配成1000μg/ml的溶液或均匀的混悬液,然后用含dmso的pbs(
‑
)稀释。最终浓度分别为:80μm、8μm、0.8μm、0.08μm、008μm。以安立生坦与伊马替尼作为对照。
[0090]
细胞增殖抑制的测试方法:96孔板每孔加入浓度为4~5
×
104个/ml的细胞悬液100μl,置37℃,5%co2培养箱内。24小时后,分别加入样品液和对照品液,10μl/孔,设双复孔,37℃,5%co2作用24小时。每孔加入5mg/ml的mtt(3
‑
(4,5
‑
二甲基噻唑
‑2‑
基)
‑
2,5
‑
二苯基四唑翁溴化物)溶液15μl,作用4小时后加入溶解液dmso,100μl/孔,置培养箱内,溶解后用mk
‑
2全自动酶标仪测490nm od值,计算抑制率。
[0091]
实验结果见表1。
[0092]
表1.样品对人体肿瘤细胞的体外增殖抑制活性ic50值。
[0093][0094]
以上实验数据显示,虽然本发明提供的苯基嘧啶胺类化合物抗肿瘤活性差别较大,但部分化合物具有极好的体外抗肿瘤活性,因而值得深入研究,以及为开发以内皮素受体为新靶点的抗肿瘤药物开辟了新的途径。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1