巴西苏木素类天然产物(+)-Brazilin的合成方法与流程
本发明涉及具有抗癌活性化合物的关键中间体的合成方法,具体而言,是一种巴西苏木素类天然产物(+)-brazilin的合成方法。
背景技术:
天然产物brazilin(巴西苏木素)是传统中药苏木中特有的一类化合物,其从20世纪50年代开始就被陆续发现,目前已分离鉴定出的主要有以下6种化合物:(+)-brazilin、(-)-brazilein、(+)-brazilidea、(+)-haematoxylin、(+)-haematoxylane以及(+)-brazilane。这类化合物具有相似的化学结构,是一类高异黄酮衍生物,都具有苯丙二氢吡喃环。brazilin的结构具有多种生物活性,因此受到了众多研究者的关注。其最早被用作天然的红色生物染色剂,随着研究的深入,化学工作者又发现其具有降血糖、抗血小板、抗炎、抗菌、抗肿瘤、保肝、抗癌等多种生物活性,甚至还是一种dna切割剂。现今,评判中药苏木的一个重要依据就是brazilin的含量。在2010年出版的《中国药典》中规定,brazilin在药材苏木中的含量不得低于0.5%。由于其独特的结构和显著的生物活性,吸引了许多化学研究者的注意并朝着合成这个分子努力。brazilin的化学结构如下:
目前文献报道的合成该天然产物的化学方法主要有:(1)heller和tamm于1980年发表在chemistryoforganicnaturalproducts上的题为fortschrittederchemieorganischernaturstoffe的研究论文;(2)kirkiacharian等人于1975年发表在bull.soc.chim.fr.上的题为hydlidereductionofcoumarinnderivatives:newmethodofsynthesisof2′‐hydroxybenzylmalonicesters的研究论文;(3)lee等人于2010年发表在bioorg.med.chem.let上的题为totalsynthesisandevaluationofbrazileinandanalogsasanti-inflammatoryandcytotoxicagents的研究论文;(4)davis,f.a等人于1993年发表在j.org.chem上的题为enantioselectivesynthesisof(+)-o-trimethylsappanoneband(+)-o-trimethylbrazilin的研究论文;(5)merlin等人于1995年发表在j.chem.soc.,perkintrans上的题为synthesisandsweetta-steofopticallyactive(-)-haematoxylinandofsome(±)-haematoxylinderivativ-es的研究论文;(5)suzuk等人于2012年发表在bioorg.med.chem.let上的题为synthesisandbiologicalevaluationofdeoxy-hematoxylinderivativesasanovelclassofanti-hiv-1agents的研究论文;(6)jahng等人于2014年发表在tetrahedron:asymmetr上的题为enantioselectivesynthesesof(+)-and(−)-brazilin的研究论文;(7)pettus等人于2005年发表在org.let上的题为synthesisof(±)-brazilinusingibx的研究论文;(8)liu等人于2008年发表在j.am.chem.soc上的题为gold-catalyzeddeoxygenativenazarovcyclizationof2,4-dien-1-alsforstereoselectivesynthesisofhighlysubstitutedcyclopentenes的研究论文;(9)zhang,h.b.等人于2011年发表在synlett上的题为designandsynthesisofbrazilin-likecompounds的研究论文;(10)zhang,h.b.等人于2013年发表在chem.commun上的题为enantioselectivetotalsynthesisof(+)-brazilin,(−)-brazileinand(+)-brazilidea的研究论文;(11)qin,h.b等人于2013年发表在tetrahedronlett.上的题为totalsynthesisof(±)-brazilinandformalsynthesisof(±)-brazilein,(±)-brazilideausingm-cpba的研究论文;(12)yadav等人于2014年发表在tetrahedron的题为formalsynthesisof(±)-brazilinandtotalsynthesisof(±)-brazilane的研究论文;(13)kim等人于2015年发表在j.org.chem上的题为totalsynthesisofbrazilin的研究论文;(14)vranken等人于2019年发表在j.org.chem上的题为totalsynthesisof(±)-brazilinusing[4+1]palladium-catalyzedcarbenylativeannulation的研究论文;2020年huang,s.p等人发表在tetrahedronlett.上的题为atotalsynthesisof(+)-brazilin的研究论文。纵观这些国内外科研工作者合成目标分子的合成路线,我们仔细分析发现,目前已通的合成路线大多都具有合成步骤较长,个别反应不易进行,试剂昂贵且毒性较大的特点。故寻求一条缩短合成步骤,原料常见易得,反应更加高效的全合成路线成了今后合成brazilin的重点研究方向。所以,寻找一条通用的、高效的、低成本的合成路线即成为了我们的迫切需求。
技术实现要素:
本发明的目的是提供一种合成步骤较少,反应更加高效的巴西苏木素类天然产物(+)brazilin的合成方法。
为解决以上技术问题,本发明采用的技术方案是:一种巴西苏木素类天然产物(+)brazilin的合成方法,反应式如下所示:
合成路线包括步骤:
步骤一,以式1化合物3,4-二甲氧基苄醇为初始原料,以二氯甲烷为溶剂,向其中加入4-二甲氨基吡啶(dmap)及对甲苯磺酰氯,再逐滴加入三乙胺,室温反应,进行对甲苯磺酰化反应得到单羟基保护产物;然后在0ºc条件下,先将氢化钠溶解在四氢呋喃以及n,n-二甲基甲酰胺溶液中,再向其中缓滴加丙二酸二乙酯,然后向反应液中滴加上述得到的单羟基保护产物,移至室温反应,获得式2化合物;
步骤二,将式2化合物溶于四氢呋喃溶剂中,向其中加入单质碘及醋酸钠,从季碳位置氧化出一个羟基,得到式3化合物;
步骤三,式3化合物在氢化铝锂的存在下,发生氢化铝锂还原反应,得到式4化合物;
步骤四,式4化合物在乙烯乙酸酯作为乙酰基供体,在liaseps酶的催化作用下发生二醇的去对称化反应,从而形成手性季碳中心,得到手性前体式5化合物;
步骤五,式5化合物在4-二甲氨基吡啶、三乙胺的作用下,与对甲苯磺酰氯发生对甲苯磺酰化反应对羟基上保护基,得到式6化合物;
步骤六,将式6化合物滴加到溶于dmf溶剂中的3-甲氧基苯酚和碳酸铯的反应体系中,发生亲核取代反应,得到式7化合物;
步骤七,将式7化合物溶解在甲醇中,再加入k2co3,进行脱除乙酰基保护的反应,得到式8化合物;
步骤八,式8化合物中加入n,n-二异丙基乙胺、二甲基亚砜和三氧化硫吡啶复合物,以二氯甲烷为溶剂,发生氧化反应得到式9化合物;
步骤九,将三氟乙酸加入到式9化合物中,以二氯甲烷为溶剂,发生一锅法分子内prins/friedel-crafts串联环化反应得到式10化合物;
步骤十,将式10化合物溶解在二氯甲烷中,经三溴化硼作用脱甲醚保护,得到产物化合物(+)brazilin;
优选地,步骤一中,在氮气的保护下,将式1化合物溶于二氯甲烷中,然后加入4-二甲氨基吡啶和对甲苯磺酰氯,再逐滴加入三乙胺,室温反应至溶液逐渐变成淡黄色,用二氯甲烷萃取,收集有机相,减压浓缩,经过分离提纯得到单羟基保护产物;在0ºc以及氮气保护的条件下,将氢化钠溶解在无水四氢呋喃及n,n-二甲基甲酰胺溶剂中,再缓慢滴加溶解在四氢呋喃中的丙二酸二乙酯溶液,0ºc下反应,再向反应体系中缓慢滴加单羟基保护产物的四氢呋喃溶液,移至室温反应后加入饱和氯化铵水溶液淬灭,用乙酸乙酯萃取后,减压浓缩,分离提纯获得式2化合物。
优选地,步骤二中,在敞口的条件下,将溶于四氢呋喃溶剂中的式2化合物、单质碘及醋酸钠于35ºc下暴露于空气中反应,反应完成后加饱和硫代硫酸钠水溶液淬灭,用二氯甲烷萃取,减压浓缩,分离提纯得到式3化合物。
优选地,步骤三中,在0ºc及氮气的保护下,将氢化铝锂溶于四氢呋喃中,再将式3化合物缓慢滴加到反应体系中,在此温度下搅拌;随后移至室温反应,然后加入甲醇、15%的氢氧化钠溶液、水以及2m的盐酸溶液淬灭,用乙酸乙酯萃取,减压浓缩,分离纯化获得式4化合物。
优选地,步骤四中,在室温以及氮气保护下,向式4化合物中加入乙酸乙烯酯,然后加入lipaseps酶,室温反应,过滤,减压浓缩,分离纯化即可得到化合物5。
优选地,步骤五中,在氮气保护及0ºc下,将4-二甲氨基吡啶、对甲苯磺酰氯及三乙胺依次加入到式5化合物的二氯甲烷溶液中,移至室温反应,加水淬灭,用二氯甲烷萃取,减压浓缩,分离纯化可得式6化合物。
优选地,步骤六中,在氮气保护下,将碳酸铯及3-甲氧基苯酚溶解在dmf溶剂中,于60ºc加热反应,再将式6化合物逐滴滴加到反应体系中,继续升温至85ºc反应,加入水溶液淬灭,用乙酸乙酯萃取后,减压浓缩,分离提纯可得式7化合物。
优选地,步骤七中,室温下,式7化合物的甲醇溶液中加入碳酸钾,反应完成后减压浓缩,分离纯化获得式8化合物。
优选地,步骤八中,0ºc及氮气保护下,将n,n-二异丙基乙胺、二甲基亚砜、三氧化硫吡啶复合物依次加入到式8化合物的无水二氯甲烷溶液中,0ºc下反应,然后用二氯甲烷萃取后,减压浓缩,分离提纯获得式9化合物。
优选地,步骤九中,将式9化合物溶解在二氯甲烷中,在氮气保护下,向上述溶液中滴加三氟乙酸于室温反应后减压浓缩,分离纯化即获得式10化合物。
优选地,步骤十中,将式10化合物溶解在二氯甲烷中,降温至-78ºc,将三溴化硼滴加到溶于化合物10的无水二氯甲烷溶液中,在-78ºc下反应后移至室温继续反应,得到产物(+)brazilin。
本发明利用lipaseps酶催化反应,mitsunobu反应,戴斯-马丁氧化反应以及在三氟乙酸(tfa)催化下发生分子内prins/friedel-crafts串联反应等成功实现了天然产物brazilin的不对称合成。
本发明的整条合成路线设计新颖独特,使用试剂均为常规化学试剂,反应条件温和,操作简单,速率相对较快,反应副产物较少,其使合成步骤大大缩短,从而在很大程度上降低了合成成本。
附图说明
图1至图12分别为下列化合物的nmr图谱。图1为式2化合物-1h,图2为式3化合物-1h,图3为式4化合物-1h,图4为式5化合物-1h,图5为式7化合物-1h,图6为式7化合物-13c,图7为式8化合物-1h,图8为式8化合物-13c,图9为式9化合物-1h,图10为式9化合物-13c,图11为式10化合物-1h,图12为式10化合物-13c,图13为(+)-brazilin-1h,图14为(+)-brazilin-13c。
具体实施方式
本发明一种典型的实施方式提供的巴西苏木素类天然产物(+)brazilin的合成方法,其反应式如下:
根据以上反应式,本发明提供一条全新的合成天然产物brazilin的合成路线,以式1化合物3,4-二甲氧基苄醇为初始原料,通过亲核取代反应,氢化铝锂还原反应,lipaseps酶催化的去对称化反应,mitsunobu反应,戴斯-马丁氧化反应以及在酸性条件下发生一锅法prins/friedel-crafts串联反应等来合成天然产物(+)brazilin;上述合成方法的反应步骤为:
步骤一,以式1化合物3,4-二甲氧基苄醇为初始原料,二氯甲烷为溶剂,三乙胺为缚酸剂,经过对甲苯磺酰氯对羟基上保护基后与氢化钠及丙二酸二乙酯反应,得到式2化合物。
步骤二,在单质碘及醋酸钠的作用下,式2化合物经过氧化得到式3化合物。相对具体地,是将式2化合物溶于四氢呋喃溶剂中,向其中加入单质碘及醋酸钠,从季碳位置氧化出一个羟基,得到式3化合物。
步骤三,式3化合物在氢化铝锂的存在下,发生氢化铝锂还原反应,得到式4化合物;
步骤四,式4化合物在乙烯乙酸酯作为乙酰基供体,在liaseps酶的催化作用下发生二醇的去对称化反应,从而形成手性季碳中心,得到手性前体式5化合物;
步骤五,式5化合物在4-二甲氨基吡啶、三乙胺的作用下,与对甲苯磺酰氯发生对甲苯磺酰化反应对羟基上保护基,得到式6化合物;
步骤六,将式6化合物滴加到溶于dmf溶剂中的3-甲氧基苯酚和碳酸铯的反应体系中,发生亲核取代反应,得到式7化合物;
步骤七,将式7化合物溶解在甲醇中,再加入k2co3,进行脱除乙酰基保护的反应,得到式8化合物;
步骤八,式8化合物中加入n,n-二异丙基乙胺、二甲基亚砜和三氧化硫吡啶复合物,以二氯甲烷为溶剂,发生氧化反应得到式9化合物;
步骤九,将三氟乙酸加入到式9化合物中,以二氯甲烷为溶剂,发生一锅法分子内prins/friedel-crafts串联环化反应得到式10化合物;
步骤十,将式10化合物溶解在二氯甲烷中,经三溴化硼作用脱甲醚保护,得到产物化合物(+)brazilin。
本发明提供的整条路线设计新颖独特,实现了天然产物brazilin的单一选择性合成,反应速率相对较快,副产物较少,反应产率高;使用的试剂均为常规化学试剂,廉价易得,使生产成本大幅降低;整个反应条件温和,操作工艺简单,适合用于工业生产。
下面通过一些实施例对本发明要求保护的技术方案作进一步说明。但是,实施例是用于解释本发明实施方案,并不超出本发明主题的范围,本发明保护范围不受所述实施例的限定。除非另作特殊说明,本发明中所用材料、试剂均可从本领域商业化产品中获得。
式2化合物的合成:
在氮气保护下,将式1化合物(0.2g,1.19mmol)溶于二氯甲烷(5ml)中,依次加入4-二甲氨基吡啶(0.0872g,0.71mmol)及对甲苯磺酰氯(0.2720g,1.43mmol),再加入三乙胺(0.17ml,1.19mmol)。于室温反应2小时,待tlc检测反应完成后,体系用水淬灭,用二氯甲烷(3×6ml)萃取,收集有机相,无水na2so4干燥,过滤除去固体杂质,减压蒸馏除去有机溶剂,经柱层析分离提纯产物(乙酸乙酯:石油醚1:10),最后洗脱液浓缩得到淡黄色油状单羟基保护产物。收率:83%。
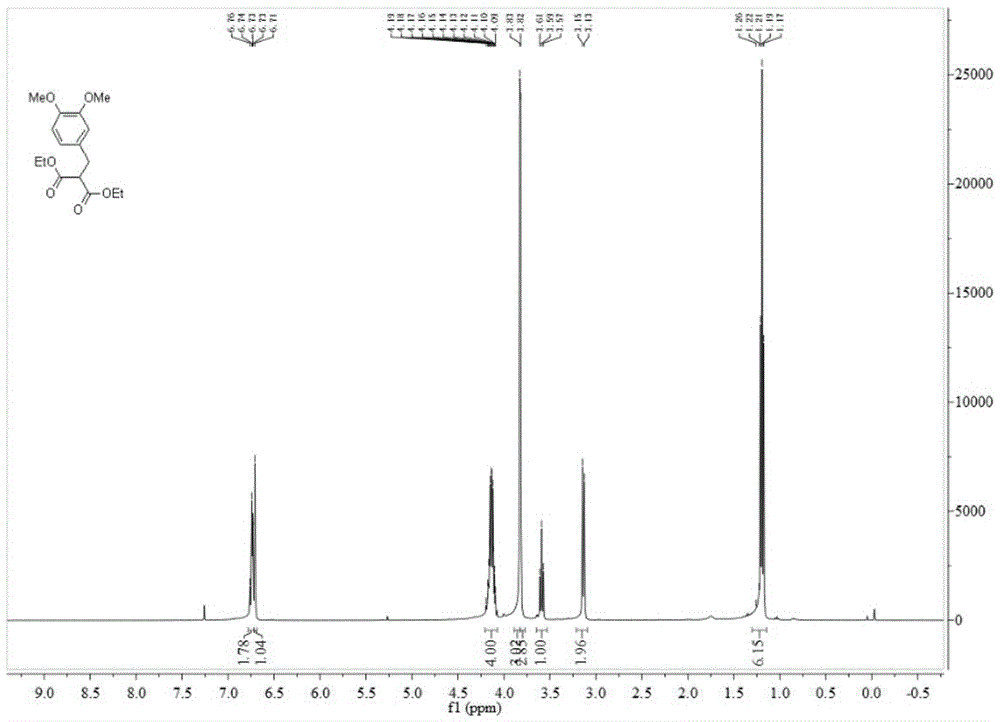
在0ºc及氮气保护下,将氢化钠(60%inmineraloil;0.0513g,1.28mmol)溶于无水四氢呋喃(2ml)与dmf(2ml)中,向反应体系中逐滴滴加溶于四氢呋喃(2ml)中的丙二酸二乙酯(0.24ml,1.60mmol),于0ºc下搅拌30分钟,接着将溶于四氢呋喃(2ml)中的单羟基保护产物(0.34g,1.07mmol)逐滴滴加到反应体系中,随后转移至室温反应约12小时,待tlc检测反应完成后,将反应液冷却至0ºc,用饱和氯化铵溶液淬灭,乙酸乙酯(3×5ml)萃取,合并有机相,无水na2so4干燥,过滤除去固体杂质,减压蒸馏后用柱层析分离提纯产物(乙酸乙酯:石油醚1:15),最后洗脱液浓缩得无色油状的式2化合物(0.3g)。收率:90%。1hnmr(400mhz,cdcl3):δ6.74(dd,j=8.7,4.9hz,2h),6.71(s,1h),4.19-4.09(m,4h),3.83(s,3h),3.82(s,3h),3.60(dd,j=17.0,9.2hz,1h),3.14(d,j=7.8hz,2h),1.30–1.14(m,6h)。
式3化合物的合成:
将式2化合物(0.1g,0.32mmol)溶解在四氢呋喃(3ml)溶剂中,再向其中依次加入单质碘(0.082g,0.32mmol)、醋酸钠(0.044g,0.32mmol),升温至35ºc,暴露于空气中搅拌反应48小时。待tlc检测反应完成后,加入饱和硫代硫酸钠水溶液淬灭反应,用二氯甲烷(3×5ml)萃取,收集有机相,用无水na2so4进行干燥,减压旋走有机溶剂,粗产品经柱层析分体提纯产物(乙酸乙酯:石油醚=1:4),洗脱液经浓缩得到无色油状的式3化合物(0.1g)。收率:98%。1hnmr(400mhz,cdcl3)δ6.80(s,1h),6.75(s,2h),4.23(q,j=6.8hz,4h),3.83(s,6h),3.74(s,1h),3.28(s,2h),1.27(t,j=7.1hz,6h)。
式4化合物的合成:
在0ºc及氮气的保护下,将氢化铝锂(0.048g,1.23mmol)溶于无水四氢呋喃(3ml),再将溶于四氢呋喃(3ml)的化合物3(0.2g,0.61mmol)滴加到其中,于0ºc下搅拌30分钟,后移至室温反应4小时,待tlc检测反应完成后,将反应体系冷却至0ºc,用甲醇、15%的氢氧化钠溶液、水以及2m的盐酸溶液淬灭,乙酸乙酯(3×10ml)萃取,无水na2so4干燥,过滤除去固体杂质,减压蒸馏后用柱层析分离提纯产物(乙酸乙酯:石油醚=2:1),最后洗脱液浓缩得无色油状的式4化合物(0.094g)。收率:63%。1hnmr(400mhz,cdcl3)δ6.80(dd,j=4.9,3.1hz,2h),6.76(dd,j=8.2,1.7hz,1h),3.87(s,3h),3.86(s,3h),3.57(q,j=11.2hz,4h),2.74(s,2h),2.55(s,2h),1.81(s,1h)。
式5化合物的合成:
在室温下,将式4化合物(0.32g,1.32mmol)溶于乙酸乙烯酯(12ml)中,接着向其中加入lipaseps酶(0.32g),室温反应6小时,待tlc检测反应完成后,过滤除去酶,减压蒸馏后用柱层析分离提纯产物(乙酸乙酯:石油醚=1:1),最后洗脱液浓缩得淡黄色液体即为式5化合物(0.587g)。收率:95%,er值为2.6:1。hplc{shimadzulc-10avp,hexane/2-propanol=7:3(v/v)}.1hnmr(400mhz,cdcl3)δ6.81(dd,j=8.6,4.9hz,2h),6.76(dd,j=8.1,1.9hz,1h),4.08(d,j=11.5hz,1h),4.00(d,j=11.5hz,1h),3.87(s,3h),3.87(s,3h),3.47(q,j=11.5hz,2h),2.79(s,2h),2.39(s,1h),2.14(s,3h)。
式6化合物及式7化合物的合成:
在氮气保护下,将式5化合物(0.3g,1.06mmol)溶解于二氯甲烷(5ml)溶剂中,降温至0ºc,依次加入4-二甲氨基吡啶dmap(0.064g,0.53mmol)、对甲苯磺酰氯(0.2414g,1.27mmol)及三乙胺(0.3ml,2.11mmol),移至室温搅拌反应10小时,tlc监测反应完成后,加水淬灭反应,二氯甲烷(3×8ml)萃取,合并有机相,无水na2so4进行干燥,减压旋走有机溶剂,粗产品经柱层析分离提纯产物(乙酸乙酯:石油醚=1:2),收集含有目标产物的洗脱液,减压浓缩得到淡黄色油状的式6化合物(0.35g)。收率:75%。
将3-甲氧基苯酚(0.025ml,0.23mmol)及碳酸铯(0.744g,2.3mmol)溶解到n,n-二甲基甲酰胺(1ml)溶剂中,升温至60ºc,搅拌反应30分钟后,将化合物6(0.1g,0.23mmol)加入到其中,继续升温至85ºc,搅拌反应30分钟。tlc监测反应完成后,加水淬灭反应,用乙酸乙酯(3×5ml)萃取,合并有机相,用饱和nacl水溶液进行洗涤,无水na2so4进行干燥,减压旋走有机溶剂,得到的粗产品经分离提纯产物(乙酸乙酯:石油醚=1:10),洗脱液浓缩得到无色液体,即为式7化合物(0.055g)。收率:68%。1hnmr(400mhz,cdcl3)δ7.18(t,j=8.2hz,1h),6.80–6.72(m,3h),6.55–6.47(m,3h),3.85(s,3h),3.82(s,1h),3.79(s,3h),3.74(dd,j=12.2,7.5hz,2h),3.67(s,1h),3.65(s,3h),2.99–2.88(m,2h),1.63(s,3h).;13cnmr(100mhz,cdcl3):δ171.23,161.01,160.25,149.00,147.64,131.57,130.07,121.26,112.42,111.36,106.85,106.53,101.13,66.65,64.48,56.05,55.85,55.45,40.15,34.15,21.13。
式8化合物的合成:
室温下,将式7化合物(0.2746g,0.70mmol)溶于甲醇(4ml)中,再加入碳酸钾(0.1457g,1.06mmol),于室温搅拌反应4小时,待tlc检测反应完成后,加入水淬灭反应,乙酸乙酯(3×5ml)萃取,减压蒸馏后用柱层析分离提纯产物(乙酸乙酯:石油醚=1:3),最后洗脱液浓缩得淡黄色液体即为式8化合物(0.2132g)。收率:87%。1hnmr(400mhz,cdcl3)δ7.11(t,j=8.4hz,1h),6.69(d,j=7.4hz,3h),6.46(dd,j=13.6,6.8hz,3h),3.77(s,3h),3.74(s,1h),3.70(s,3h),3.69–3.64(m,2h),3.59(s,1h),3.55(s,3h),2.87(q,j=13.6hz,2h).13cnmr(100mhz,cdcl3)δ160.73,159.46,148.54,147.65,129.84,128.43,122.29,113.59,111.05,106.60,100.99,74.14,68.85,65.526,55.65,55.32,55.06,39.60.hrms(esi):m/zcalcd.forc19h25o6,[m+h]+349.1646,found349.1648。
式9化合物的合成:
在氮气保护下,将式8化合物(2.0g,5.75mmol)溶于无水二氯甲烷(30ml)中,降温至0ºc后,依次加入n,n-二异丙基乙胺(2.23g,17.24mmol,3eq.)、二甲基亚砜(4.49g,57.47mmol,10eq.)和三氧化硫吡啶复合物(2.75g,17.24mmol,3eq.),在此温度下搅拌反应1小时。tlc检测当原料完全反应后,加入稀盐酸溶液中和过量的n,n-二异丙基乙胺,二氯甲烷萃取(20ml×3),合并所得有机相,饱和食盐水洗涤,再用无水na2so4干燥。抽滤去除固体杂质,减压浓缩后粗产物经柱层析分离提纯(石油醚:乙酸乙酯=8:1),将所得到含有目标产物的洗脱液经减压浓缩后得到了无色粘稠液体即为式9化合物(1.81g)。收率:91%。1hnmr(400mhz,cdcl3)δ9.77(s,1h),7.13(t,j=8.2hz,1h),6.73(dd,j=9.6,7.6hz,3h),6.57–6.48(m,1h),6.43(d,j=7.1hz,2h),4.10(d,j=9.4hz,1h),3.89(d,j=9.4hz,1h),3.80(s,3h),3.73(d,j=6.1hz,6h),3.61(s,1h),3.02(s,2h).13cnmr(100mhz,cdcl3)δ203.248,161.31,159.73,149.24,148.69,130.42,126.84,122.88,114.16,111.66,107.67,107.06,101.68,79.95,70.77,56.23,56.16,55.66,39.24.hrms(esi):m/zcalcd.forc19h23o6,[m+h]+347.1489,found347.1492。
式化合物10的合成:
在氮气保护下,将式9化合物(1.81g,5.23mmol)溶于无水二氯甲烷(50ml)中,然后将三氟乙酸(2.40g,21.40mmol,4eq.)逐滴加入到反应体系中,搅拌反应1小时。tlc检测反应完成后,加入适量的饱和碳酸氢钠溶液淬灭反应,二氯甲烷萃取(20ml×3),合并所得有机相,饱和食盐水洗涤,再用无水na2so4干燥除去有机相中的水分。抽滤,减压浓缩得到的粗产品经硅胶柱分离提纯(石油醚:乙酸乙酯=6:1),洗脱液减压浓缩后得到白色固体式10化合物(1.42g)。收率:83%。1hnmr(400mhz,cdcl3)δ7.27(d,j=8.4hz,1h),6.78(s,1h),6.73(s,1h),6.64(dd,j=8.4,2.3hz,1h),6.44(d,j=2.3hz,1h),4.07(s,1h),4.01(d,j=11.3hz,1h),3.83(d,j=3.9hz,6h),3.79(d,j=11.3hz,1h),3.74(s,3h),3.21(d,j=15.8hz,1h),2.85(d,j=15.8hz,1h).13cnmr(100mhz,cdcl3)δ159.40,154.38,148.73,148.45,136.19,130.65,114.46,108.84,108.52,107.80,101.98,77.45,70.26,56.12,56.08,55.32,50.48,41.43.hrms(esi):m/zcalcd.forc19h21o5[m+h]+329.1384,found329.1385。
(+)-brazilin的合成:
氮气保护下,将化合物10(1.42g,4.34mmol)溶于无水二氯甲烷(85ml)中,降温至-78ºc,将三溴化硼(21.7ml,21.70mmol,1minch2cl2,5eq.)逐滴滴加到反应体系中,在-78℃下反应2小时后移至室温反应16小时。tlc检测反应完全后,加入水淬灭反应,减压浓缩除去二氯甲烷后,用乙酸乙酯萃取(30ml×3),合并所得有机相,饱和食盐水洗涤,再用无水na2so4干燥除去溶液中水分。抽滤去除固体杂质,减压浓缩得到的粗产品经柱层析分离提纯(石油醚:乙酸乙酯=1:1),将所得到含有目标产物的洗脱液经减压浓缩后得到了琥珀色固体产物(1.03g)。收率:83%。1hnmr(400mhz,cd3od)δ7.16(d,j=8.3hz,1h),6.71(s,1h),6.60(s,1h),6.47(dd,j=8.3,2.3hz,1h),6.31(d,j=2.3hz,1h),3.96(s,1h),3.92(d,j=11.5hz,1h),3.67(d,j=11.3hz,1h),3.01(d,j=15.6hz,1h),2.75(d,j=15.7hz,1h).13cnmr(100mhz,cd3od)δ157.73,155.68,145.57,145.25,137.51,132.29,131.46,115.63,113.00,112.53,110.06,104.35,78.15,70.86,51.05,49.96,42.86.hrms(esi):m/zcalcd.forc16h15o5,[m+h]+287.0914,found287.0914。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!