一种银催化合成异香豆素类衍生物的方法及其应用
本发明属于有机合成化学技术领域,具体涉及一种银催化合成异香豆素类衍生物的方法及其应用。
背景技术:
公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
异香豆素的衍生物是有机合成中有价值的合成中间体,在许多天然产物和药物中都有其特点。2012年,尹立等人研究发现异香豆素对患有老年痴呆症的小白鼠有一定的治疗作用;王知斌等人在2012年研究发现从八仙花中提取了两种异香豆素类化合物绣球酚和绣球酚-4’-o-β-d-葡萄糖苷,并报道了两种异香豆素类化合物具有降血糖和护肝的功能;日本学者johji于1994年发现八仙花中的异香豆素类化合物具有抗溃疡、抗过敏等生物活性。一般地,天然产物中提取出来的具有生物活性的异香豆素衍生物活性成分往往较低,为了增强所得化合物的生物活性,需要对其结构进行优化和改造,人们主要借助过渡金属催化建立了各种合成方法来获得这些杂环化合物。π-亲电过渡金属催化剂介导的炔基取代的羧酸或酯的分子内环化是构建异香豆素衍生物的常见策略之一,通常此类方案依赖于通过π的配位活化炔丙基c≡c键-亲电过渡金属催化剂,该催化剂可引发6-内杂环反应并产生结构多样的六元杂环化合物。异香豆素支架的另一种方法是通过金属盐介导的环化作用自发形成碳-金属键和杂环骨架,并通过适当的合成反应(如交叉偶联和α-芳基化)生成有机金属中间体,从而产生目标杂环。这些策略可直接可以从相对简单的有机底物中获得各种五元和六元杂环。
邻炔基苯甲醛构成多方面的和容易获得的构建块,适用于各种杂环化合物的构建化合物.杂环化邻炔基苯甲醛的合成为取代异香豆素的合成提供了一条方便的途径;然而,迄今为止,只有少数相关的合成方案被披露。值得注意的是,youn及其同事报导了nhc催化的邻炔基苯甲醛氧化分子内杂环化以生成五元和六元杂环支架的开创性工作。不幸的是,该方案提供了具有低选择性的相应产物,其高度依赖于起始邻炔基苯甲醛中取代基的性质。随后,singh和ghorai课题组扩大了邻炔基吡啶醛杂环化的底物范围,实现了吡喃并[4,3-b]喹啉酮的选择性形成。关于结构相关的邻炔基苯甲醛,迄今为止仅实现了其杂环化的一个实例。
在本发明人在研究过程中发现,这些合成异香豆素类衍生物方法中存在以下问题:1.催化剂价格昂贵,合成成本高;2.无现成底物,需要进一步合成,步骤繁琐复杂;3.区域选择性较差;4.反应产率不高且副产物较多,影响提纯。
技术实现要素:
为了解决现有技术的不足,本发明提供一种银催化合成异香豆素类衍生物的方法及其应用,该方法能够直接利用2-(苯乙炔基)苯甲醛为模型底物一步合成异香豆素类衍生物,无需强氧化剂,没有毒副产物,更适宜大规模的工业化生产。
为了实现上述目的,本发明第一方面提供一种银催化合成异香豆素类衍生物的方法,以2-(苯乙炔基)苯甲醛为模型底物,以一价银盐作为催化剂,在不低于30℃的温度下进行反应,生成一种异香豆素类衍生物,其结构式为:
其中,2-(苯乙炔基)苯甲醛的化学式为
反应式为:
所述一价银盐为agbf4、agno3、ag2co3、agotf中的一种。溶剂为ch3cn、dmf、甲苯、dce、dma中的一种。
本发明第二方面提供上述方法在制备医疗保健型药物中的应用。
本发明的一个或多个实施方式至少具有以下有益效果:
本发明提供了一种银催化合成异香豆素类衍生物的方法,首次以2-(苯乙炔基)苯甲醛为模型底物,在一价银盐的催化作用下一步制备异香豆素衍生物。该方法简便、高效,所用到的原料简单易得且无毒、步骤少、条件温和、成本低,适宜大规模的工业化生产。
附图说明
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
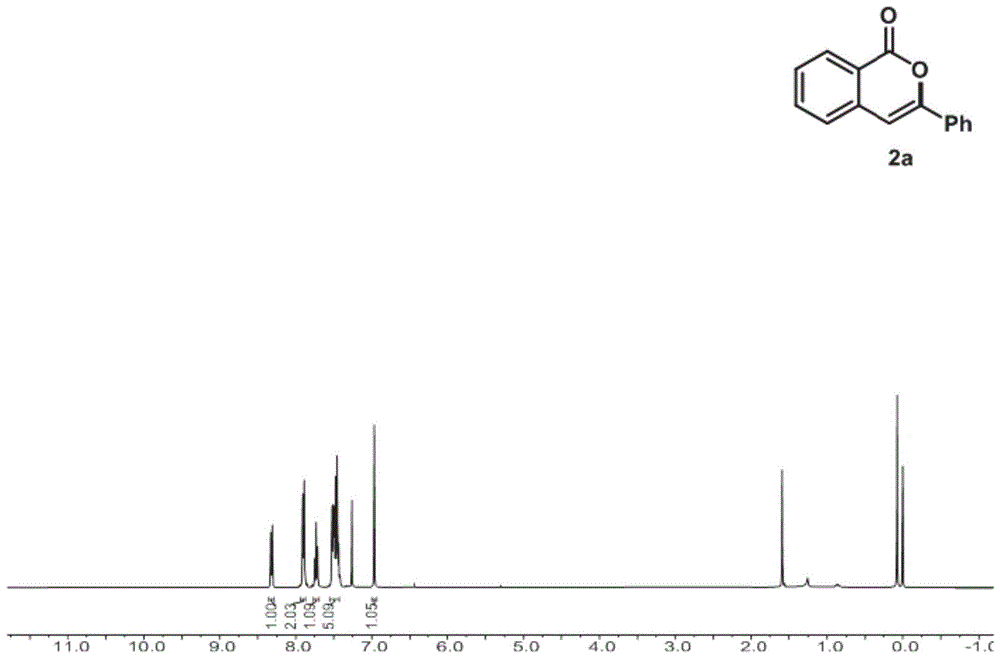
图1为本发明实施例2制备的化合物2a的1h-nmr的核磁共振谱图;
图2为本发明实施例2制备的化合物2a的13c-nmr的核磁共振谱图;
图3为本发明实施例5制备的化合物2b的1h-nmr的核磁共振谱图;
图4为本发明实施例5制备的化合物2b的13c-nmr的核磁共振谱图;
图5为本发明实施例6制备的化合物2c的1h-nmr的核磁共振谱图;
图6为本发明实施例6制备的化合物2c的13c-nmr的核磁共振谱图;
图7为本发明实施例7制备的化合物2d的1h-nmr的核磁共振谱图;
图8为本发明实施例7制备的化合物2d的13c-nmr的核磁共振谱图。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
正如背景技术所介绍的,现有技术中异香豆素类衍生物合成方法存在底物不易获取、反应条件苛刻、有毒副产物、合成成本高等问题,为了解决如上的技术问题,本发明第一方面提出了一种银催化合成异香豆素类衍生物的方法,以2-(苯乙炔基)苯甲醛为模型底物,以一价银盐作为催化剂,在不低于30℃的温度下进行反应,生成一种异香豆素类衍生物,其结构式为:
其中,2-(苯乙炔基)苯甲醛的化学式为
反应式为:
在本发明的一个或多个实施方式中,所述一价银盐为agbf4、agno3、ag2co3、agotf中的一种;
优选的,所述一价银盐为四氟硼酸银或硝酸银;
进一步优选的,所述一价银盐为四氟硼酸银。
该催化剂能够提高原料的转化率和产物的产率,当一价银盐为四氟硼酸银时,能够进一步提高异香豆素衍生物的产率。
在本发明的一个或多个实施方式中,r选自氢、4-氟苯基或4-甲基苯基;
r1选自c1-c5直链烷基或烷氧基、芳基、取代芳基、杂环基、取代杂环基、芳杂基、取代芳杂基,优选为芳基或取代芳基。
在本发明的一个或多个实施方式中,所述芳基为苯基,所述给电子或吸电子的取代芳基为被卤素、c1-c6直链的烷基、c1-c6支链的烷基、c1-c2直链的烷氧基或c1-c2支链的烷氧基所取代的苯基,所述芳杂基中含有一个或多个杂原子,杂原子选自n、o、s。
在本发明的一个或多个实施方式中,所述给电子或吸电子的取代芳基为被f、cl、br、甲基、乙基、正丙基、叔丁基、正戊基、甲氧基、乙氧基等所取代的苯基;
在本发明的一个或多个实施方式中,所述反应温度为30~60℃,该温度能够提高原料的转化率,同时提高产物的产率。
优选的,反应的温度为40±8℃时,能够进一步提高原料的转化率和产物的产率。
在本发明的一个或多个实施方式中,将原料添加至溶剂中溶解,再加入催化剂,加热进行反应。溶剂能提高原料的转化率,同时提高产物的产率。
所述溶剂选自ch3cn、dmf、甲苯、dce、dma的一种或多种。
优选的,溶剂为n,n-二甲基甲酰胺(dmf)或甲苯。
进一步优选的,溶剂为甲苯。
在本发明的一个或多个实施方式中,2-(苯乙炔基)苯甲醛与一价银盐的摩尔比为10:1。
在本发明的一个或多个实施方式中,反应时间为0~10h,且反应时间不为0。
优选的,反应时间为6±0.5h。
在本发明的一个或多个实施方式中,将反应后溶液加入萃取溶剂进行萃取获得有机相,将有机相中的溶剂去除,进行硅胶柱层析,获得异香豆素衍生物。
萃取采用的萃取溶剂为1,2-二氯乙烷、甲苯、硝基甲烷、乙酸乙酯、乙醚、正己烷、环己烷、石油醚或二氯甲烷中的一种或多种;
萃取采用的萃取溶剂为二氯甲烷;
所述萃取进行1~3次,每次使用5~20ml萃取溶剂;
获得有机相采用无水硫酸镁进行干燥,再去除有机溶剂。
硅胶柱层析的洗脱液为石油醚和乙酸乙酯;
石油醚和乙酸乙酯的体积比为1~50:1~3,优选为20:1。
本发明第二方面提供上述方法在制备医疗保健型药物中的应用。
为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
实施例1
将化合物1a即2-(苯乙炔基)苯甲醛(0.0560mg,0.5mmol)加入到2ml的dmf中,在40℃的条件下溶解,随后向体系中加入四氟硼酸银(0.0097g,0.05mmol),继续加热搅拌6小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=100:5)得到化合物2a的产率为75%。
实施例2
将化合物1a即2-(苯乙炔基)苯甲醛(0.0560mg,0.5mmol)加入到2ml的甲苯中,在40℃的条件下溶解,随后向体系中加入四氟硼酸银(0.0097g,0.05mmol),继续加热搅拌6小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=100:5)得到化合物2a的产率为88%。
实施例3
将化合物1a即2-(苯乙炔基)苯甲醛(0.0560mg,0.5mmol)加加入到2ml的甲苯中,在40℃的条件下溶解,随后向体系中加入硝酸银(0.0085g,0.05mmol),继续加热搅拌6小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=100:5)得到化合物2a的产率为60%。
实施例4
将化合物1a即2-(苯乙炔基)苯甲醛(0.0560mg,0.5mmol)加入到2ml的甲苯中,在40℃的条件下溶解,随后向体系中加入碳酸银(0.0140g,0.05mmol),继续加热搅拌6小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=100:5)得到化合物2a的产率为78%。
实施例1~4的反应如下式所示:
化合物1a:
1hnmr(400mhz,cdcl3),如图1所示,δ6.97(s,1h,ch),7.43–7.53(m,5h,arh),7.71–7.75(m,1h,arh),7.88–7.91(m,2h,arh),8.32(d,j=8.0hz,1h,arh),13c-nmr(cdcl3,100mhz),如图2所示,δ76.7,101.8,120.6,125.3,126.0,128.2,128.9,129.7,130.0,132.0,134.9,137.5,153.7,162.4;hrms(esi)m/zcalculatedforc15h11o2[m+h]+:223.0754,found223.0760.
实施例5
将化合物1b即5-氟-2-(苯乙炔基)苯甲醛(0.0540mg,0.5mmol)加入到2ml的甲苯中,在40℃的条件下溶解,随后向体系中加入四氟硼酸银(0.0097g,0.05mmol),继续加热搅拌6小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=100:5)得到化合物2b的产率为71%。
化合物2b:
1hnmr(400mhz,cdcl3),如图3所示,δ6.91(s,1h,ch),7.13–7.21(m,2h,arh),7.45–7.50(m,3h,arh),7.87–7.89(m,2h,arh),8.33(dd,j=8.8hz,j’=5.6hz,1h,arh);13c-nmr(100mhz,cdcl3),如图4所示,δ94.3(d,jf-c=4.8hz),120.2(d,jf-c=19.8hz),121.9(d,jf-c=4.0hz),125.3,125.4,126.7(d,jf-c=16.7hz),128.4(d,jf-c=7.6hz),128.9,130.3,131.7,154.3;hrms(esi)m/zcalculatedforc15h10fo2[m+h]+:241.0659,found241.0663.
实施例6
将化合物1c即5-甲基-2-(苯乙炔基)苯甲醛(0.0550mg,0.5mmol)加入到2ml的甲苯中,在40℃的条件下溶解,随后向体系中加入四氟硼酸银(0.0097g,0.05mmol),继续加热搅拌6小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=100:5)得到化合物2c的产率为78%。
反应如下式所示:
化合物2c:
1hnmr(400mhz,cdcl3),如图5所示,δ2.42(s,3h,ch3),6.87(s,1h,ch),7.35(d,j=8.0hz,1h,arh),7.38–7.44(m,3h,arh),7.48(dd,j=8.0hz,j’=2.0hz,1h,arh),7.80–7.83(m,2h,arh),8.06(s,1h,arh);13c-nmr(cdcl3,100mhz),如图6所示,δ21.4,101.8,120.4,125.1,125.9,128.8,129.4,129.8,132.1,135.1,136.2,138.5,152.8,162.5;hrms(esi)m/zcalculatedforc16h13o2[m+h]+:237.0910,found237.0905
实施例7
将化合物1d即2-(对甲基苯乙炔基)苯甲醛(0.0590ml,0.5mmol)加入到2ml的甲苯中,在40℃的条件下溶解,随后向体系中加入四氟硼酸银(0.0097g,0.05mmol),继续加热搅拌6小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=100:5)得到化合物2d的产率为81%。
反应如下式所示:
化合物2d:
1hnmr(400mhz,cdcl3),如图7所示,δ2.39(s,3h,ch3),6.89(s,1h,ch),7.24–7.26(m,2h,arh),7.44–7.48(m,2h,arh),7.67–7.71(m,1h,arh),7.75–7.77(m,2h,arh),8.28(d,j=8.0hz,1h,arh);13c-nmr(100mhz,cdcl3),如图8所示,δ17.1,101.1,120.4,125.2,125.8,127.9,129.2,129.5,129.6,134.8,137.7,140.3,153.8,162.4;hrms(esi)m/zcalculatedforc16h13o2[m+h]+:237.0910,found237.0911.
对比例
将化合物1e即2-(苯乙炔基)苯甲酸(0.0650mg,0.5mmol)加入到2ml的甲苯中,在40℃的条件下溶解,随后向体系中加入四氟硼酸银(0.0097g,0.05mmol),继续加热搅拌6小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=100:5)得到化合物2e的产率为29%。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!