儿茶酚1,2-双加氧酶及其编码基团、制备方法和应用
1.本发明属于生物技术领域,尤其涉及一种儿茶酚1,2-双加氧酶及其编码基团、制备方法和应用。
背景技术:
2.随着原油及其加工品使用量的增加,常规油气资源的不断消耗,重油在我国工业中所占的比例日益增大,而由重油导致的环境污染事件也呈逐年加重的态势。微生物修复技术因具有绿色、高效、经济、无二次污染等优势,被视为重油污染治理的理想技术手段。由于重油中富含难生物降解的高毒性多环芳烃组分,限制了微生物修复技术在重油污染修复中的应用。因此,如何高效去除重油中的多环芳烃组分是提高重油污染微生物修复效率的关键。
3.酶修复因具有可专一性高效降解某类污染物,且降解过程不受跨膜运输的限制,对环境营养要求不高、不易受捕食和有毒物质影响等优势,已被用于环境污染治理,因此,利用特定的酶来降解重油中的多环芳烃组分对提高重油污染物的微生物修复效率至关重要。多环芳烃生物降解的关键限速步骤是苯环的起始加氧反应和苯环的最终开环反应,利用催化氧化上述反应的酶直接降解重油中的多环芳烃组分均可提高重油的微生物降解速率。因此,开发能够高效催化苯环彻底开环的儿茶酚1,2-双加氧酶具有重要的意义。
4.儿茶酚1,2-双加氧酶是内二元醇双加氧酶,它通过将氧分子的两个氧原子插入到底物的两个碳原子上,引起苯环结构不稳定而开环,进而生成顺,顺-黏糠酸。儿茶酚1,2-双加氧酶是邻位氧化开环酶,它催化儿茶酚的邻位降解。然而,由于野生菌株的蛋白表达过程受到多种因素的调控,产酶量往往难以达到较高的水平,限制了酶制剂在污染修复领域中的应用限制。因此,利用基因工程技术克隆儿茶酚1,2-双加氧酶的编码基因,并使这些基因在异源受体上大量表达具有重要意义。
技术实现要素:
5.本发明实施例的目的在于提供一种儿茶酚1,2-双加氧酶,旨在解决背景技术中提出的问题。
6.本发明实施例是这样实现的,一种儿茶酚1,2-双加氧酶,其氨基酸序列如序列表seq id no.2所示。
7.本发明实施例的另一目的在于提供一种儿茶酚1,2-双加氧酶的编码基因,其核苷酸序列如序列表seq id no.1所示。
8.本发明实施例的另一目的在于提供一种上述的儿茶酚1,2-双加氧酶的制备方法,其包括以下步骤:
9.特异性扩增所述儿茶酚1,2-双加氧酶的编码基因,得到扩增产物;
10.将扩增产物进行双酶切后,再连接到同样经双酶切的克隆质粒上,得到重组克隆质粒;
arcat。
33.本发明实施例的另一目的在于提供一种含有儿茶酚1,2-双加氧酶的编码基因的表达菌株。
34.进一步地,将重组表达质粒电转移到表达菌株e.colibl21(de3)中,通过lb-kan平板筛选获得儿茶酚1,2-双加氧酶表达质粒pet28a-arcat的e.colibl21(de3)。
35.本发明实施例的另一目的在于提供一种上述的儿茶酚1,2-双加氧酶在重油微生物降解中的应用。
36.本发明提供的一种儿茶酚1,2-双加氧酶的制备方法,以解决野生菌朱产酶量低的应用限制;同时将儿茶酚1,2-双加氧酶用于重油污染物的微生物降解过程,以提高重油的微生物降解速率。本发明提供的可用于重油污染物修复的儿茶酚1,2-双加氧酶制剂,可以克服重油污染微生物修复的环境适应性能差、启动速度慢和重油重质组分降解性能差的缺点;同时提供了实现该儿茶酚1,2-双加氧酶制剂高效异源表达的方法,以克服儿茶酚1,2-双加氧酶工业化应用的使用限制。
37.本发明制备了一种儿茶酚1,2-双加氧酶,获得了能够高效异源表达儿茶酚1,2-双加氧酶的基因工程菌。本发明通过酶活性检测、酶学性质分析、儿茶酚1,2-双加氧酶和细菌联合降解重油的性能分析,证明本发明的儿茶酚1,2-双加氧酶是一种可用于促进重油生物降解速率的儿茶酚1,2-双加氧酶。
38.本发明的优点在于,本发明的儿茶酚1,2-双加氧酶能用于重油污染修复,将儿茶酚1,2-双加氧酶与细菌联合用于重油污染物的生物降解,重油降解效率和重油降解速率分从60.60%、0.1038d-1
上升至60.52%,和0.1586d-1
。本发明提供的儿茶酚1,2-双加氧酶制备方法可实现儿茶酚1,2-双加氧酶高效异源表达,对于实现儿茶酚1,2-双加氧酶的工业化应用具有重大的实际意义。
附图说明
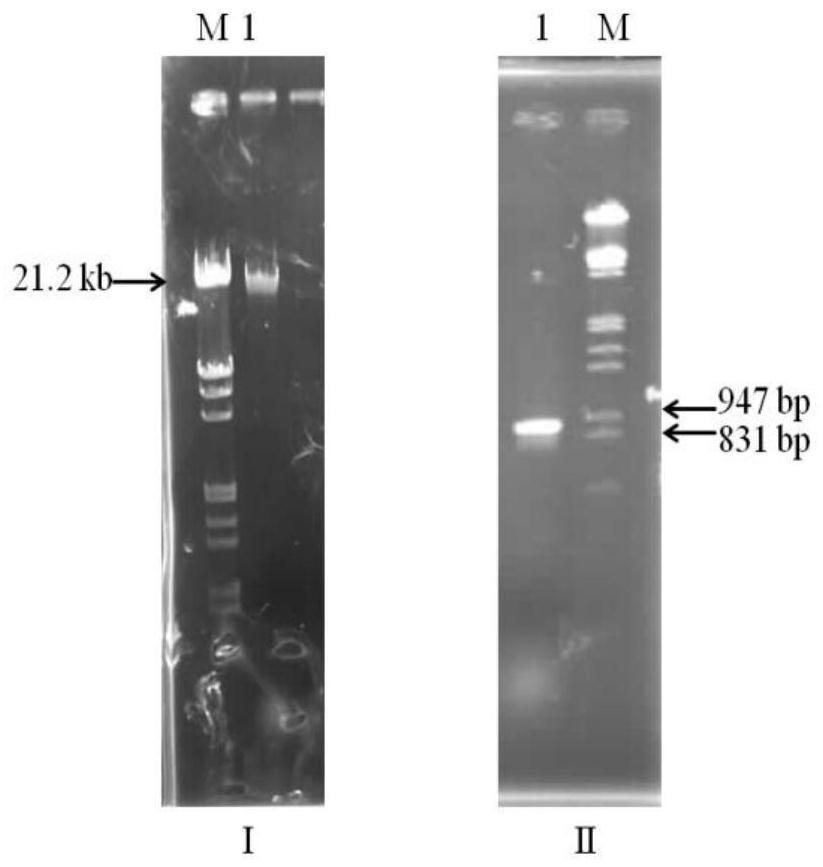
39.图1中(i)为细菌arthrobacter sp.fb24的基因组。lane m为lambda d na/ecor i+hind iii marker;lane1为arthrobacter sp.fb24基因组dna;(i i)为目的基因片段扩增;lanem为λdna/ecor i+hind iii marker;lane2为扩增获得目的基因arcat。
40.图2中(i)为lane m为λdna/ecor i+hind iii marker;lane 1为pmd19t质粒;(ii)为lane m为λdna/ecor i+hind iii marker;lane 1为pmd19t/ecor i+hind iii;(iii)为lane m为λdna/ecor i+hind iii marker;lane 1~7为pmd19t-arcat克隆pcr。
41.图3中(i)为lane m为λ-hind iii digest dna marker;lane 1~4为pmd19t-arcat;lane 5为pmd19t-arcat/ecor i+hind iii;(ii)lane m为λ-hi nd iii digest dna marker;lane 1为pet28a/ecor i+bamh i;(iii)lane m为λ-hind iii digest dna marker;lane 1为pet28a-arcat;iiii为pet28a-arcat克隆pcr鉴定。
42.图4为iptg浓度对儿茶酚1,2-双加氧酶表达的影响。lane m:marker;la ne 1~2:0.2和0.4mmol/l iptg(上清);lane 3~8:0.1~1.0mmol/l iptg。
43.图5为儿茶酚1,2-双加氧酶蛋白的纯化。lane m为marker;lane 1为诱导细胞;lane 2为细胞裂解液上清;lane 3为细胞裂解液沉淀;lane 4为ni-na t agarose吸附后残余细胞裂解液沉淀;lane 5为ni-nat agarose清洗流出液;lane 6~9为ni-nat agarose洗
脱液流出液。
44.图6为儿茶酚1,2-双加氧酶的最适反应温度及温度耐受性。
45.图7为儿茶酚1,2-双加氧酶的最适反应ph及酸碱耐受性。
46.图8为儿茶酚1,2-双加氧酶的添加对菌群dl-1314重油降解效果的影响。
47.图9为菌群dl-1314的重油降解动力学特征曲线。
48.图10为儿茶酚1.2-双加氧酶-菌群dl-1314的重油降解动力学特征曲线。
具体实施方式
49.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
50.实施例1
51.该实施例提供了一种节杆菌arthrobacter sp.fb24基因组dna的提取和儿茶酚1,2-双加氧酶编码基因序列的特异性扩增,具体如下:
52.(1)节杆菌arthrobacter sp.fb24基因组dna的提取
53.接种arthrobacter sp.fb24菌株于液体lb培养基中,37℃振荡培养过夜,8000r/min离心收集菌体。基因组提取利用omega基因组试剂盒e.z.n.a.bact erial dna kit,具体提取步骤按照试剂盒的说明进行。
54.(2)儿茶酚1,2-双加氧酶编码基因的pcr扩增
55.根据儿茶酚1,2-双加氧酶的编码基因序列(其核苷酸序列如序列表seq id no.1所示)设计合成携带限制性酶切位点ecor i和hind iii的基因扩增引物,上游引物arcatf:5
’‑
ccggaattcatgaccaagg ttccggag-3’(如序列表s eq id no.3所示),下划线表示ecori酶切位点;下游引物arcatr:5
’‑
ggga agcttt tagtcctgggggtcgag-3’(如序列表seq id no.4所示),下划线表示hind iii酶切位点。利用pcr扩增编码儿茶酚1,2-双加氧酶的基因序列a rcat。pcr扩增使用20μl体系,包括e
×
taqdna聚合酶0.3μl,10
×
pcr buf fer 2μl,dntp 0.5μl,pcr模版arthrobacter sp.z5t基因组dna 1μl,上游引物arcatf 1μl,下游引物arcatr 1μl,ddh2o 14.2μl。pcr的扩增条件为95℃预变性3min,其后30个循环,每个循环包括95℃变性45s、57℃退火45s、72℃延伸1min,循环扩增完毕后,于72℃延伸10min。
56.进一步对pcr产物进行琼脂糖凝胶电泳检测。1%琼脂糖,pcr产物上样量为5μl,电泳缓冲液为1
×
tae,100v电泳30~40min,eb染色后在紫外灯下观察结果,pcr产物分别为约841bp的电泳条带。实验结果如图1所示。
57.实施例2
58.该实施例提供了一种含儿茶酚1,2-双加氧酶编码基因的克隆菌株的构建方法,具体包括:
59.(1)酶切反应
60.将实施例1扩增获得的pcr产物和质粒pmd19t经ecor i和hind iii双酶切,酶切体系如下:1μl ecor i,1μl hind iii,5μl 10
×
buffer,载体或pc r产物500ng;ddh2o定容至50μl。将上述体系于37℃反应6h,即获得相应的酶切产物。
61.(2)重组克隆质粒pmd19t-arcat构建
62.将经过ecor i和hind iii双酶切的pcr产物连接到同样经过双酶切的pm d19t载体上,10μl连接体系如下:1μl pmd19t,5μl pcr产物,1μl 10
×
ligase buffer,1μl t4 dna ligase,2μl ddh2o,混匀,于16℃温浴16~20h取出,直接转化或放置于-20℃保存,得到第一连接产物。
63.(3)转化反应
64.将上述第一连接产物电转化至e.coli dh5α,通过lb-amp平板筛选获得c at重组克隆质粒pmd19t-arcat的e.coli dh5α克隆菌株。实验结果如图2和图3所示。
65.实施例3
66.该实施例提供了一种儿茶酚1,2-双加氧酶编码基因表达菌株的构建方法,具体包括:
67.(1)酶切反应
68.从克隆菌株中提取重组克隆质粒pmd19t-arcat,并将重组克隆质粒pmd19t-arcat和质粒pet28a经ecor i和hind iii双酶切,酶切体系如下:1μl eco r i,1μl hind iii,5μl 10
×
buffer,载体或重组克隆质粒pmd19t-arcat 500ng;ddh2o定容至50μl。将上述体系于37℃反应6h,即获得arcat基因片段。
69.(2)重组表达质粒pet28a-arcat构建
70.将经过ecor i和hind iii双酶切的arcat基因片段连接到同样经过双酶切的pet28a载体上,10μl连接体系如下:1μl pet28a、5μl arcat基因片段,1μl 10
×
ligase buffer,1μl t4 dna ligase,2μl ddh2o,混匀,于16℃温浴16~20h取出,直接转化或放置于-20℃保存,得到第二连接产物。
71.(3)转化反应
72.将上述第二连接产物电转化到e.coli bl21(de3)中,通过lb-kan平板筛选获得arcat重组表达质粒pet28a-arcat的e.coli bl21表达菌株(de3)。实验结果如图2和图3所示。
73.实施例4
74.该实施例提供了一种儿茶酚1,2-双加氧酶的表达和活性测定方法,具体包括:
75.1、儿茶酚1,2-双加氧酶的表达
76.1)将上述待表达的pet28a-arcat/e.coli bl21(de3)表达菌株划线到lb-kan平板,37℃过夜培养;
77.2)挑取单菌落于液体lb-kan中,37℃、200r/min培养过夜,次日按1%接种量转接到1000ml lb培养液中继续培养,得到培养液;
78.3)待培养液的od
600
达0.4~0.8时,加入不同浓度的(0.1,0.2,0.4,0.6,0.8,1.0mmol/l)iptg,20℃、100r/min下诱导表达5h;收集菌体,加入一定体积的细胞裂解液,置于冰上用超声波破碎,至菌液澄清时结束,即得含有儿茶酚1,2-双加氧酶(其氨基酸序列如序列表seq id no.2所示)的粗酶液;
79.4)将粗酶液经12000r/min离心15min,分别收集细胞裂解液与细胞碎片进行sds-page。实验结果表明,改变iptg的浓度,并没有明显的改变儿茶酚1,2-双加氧酶的表达量,因此,在后续的实验中以0.1mmol/l iptg的终浓度来诱导儿茶酚1,2-双加氧酶的表达,实验结果如图4所示。
80.2、儿茶酚1,2-双加氧酶活性的测定
81.以儿茶酚为氧化底物来测定儿茶酚1,2-双加氧酶。每升培养液每分钟生成1μmol儿茶酚氧化产物(顺,顺-已二烯二酸)所需的酶量为一个酶活力单位(1u)。反应体系为2ml:60μl儿茶酚(50mmol/l),60μl待测酶液,200μlna2edta(20mmol/l),1.68ml磷酸盐缓冲溶液(50mmol/l,ph=7.5),65℃下反应3min,测定反应前后体系在260nm处吸光度的变化。按照下式计算漆酶的反应活性:
82.活性(u/l)=δa/(ε
×
d)
×v总
÷v样
÷
t=661
×
δa。
83.式中:δa为吸光度的变化;ε是顺,顺-已二烯二酸的摩尔吸光系数,为16.8l/mmol/cm;d是比色皿的光径,为1cm;v
总
是反应总体积,为2ml;v
样
是待测酶液的体积,为60μl;t是反应时间,为3min。
84.实施例5
85.该实施例提供了一种儿茶酚1,2-双加氧酶的纯化方法,具体包括:
86.采用qiagen公司的ni-nta琼脂糖蛋白纯化柱纯化目标融合蛋白:
87.(1)吸取1mlni-nta溶液到15ml试管中。离心去除上清液并加入2ml裂解缓冲溶液。轻柔震荡,混匀。重复上述步骤1次;
88.(2)加入4ml已澄清的裂解液到上述已平衡的基质中,并于体系中加入一定量的咪唑,使其最终浓度为5mmol/l,于4℃摇床中轻摇(60r/min)60min;
89.(3)将“裂解物-ni-nta”混合物装入到带有底部出口的柱子中;
90.(4)取下底盖并收集流出液,保存流出液并进行电泳;
91.(5)用2.5ml的洗脱液清洗沉淀物2次,收集清洗液并进行电泳;
92.(6)用0.5ml的eb buffer洗脱蛋白质4次,收集洗脱液并于4个试管中保存,对收集的洗脱液进行电泳。实验结果如图5所示。
93.实施例6
94.该实施例提供了一种儿茶酚1,2-双加氧酶酶学性质分析方法,具体包括:
95.1、儿茶酚1,2-双加氧酶的最适反应温度及温度耐受性研究
96.1)儿茶酚1,2-双加氧酶的最适反应温度
97.将实施例4制备的儿茶酚1,2-双加氧酶粗酶液用50mmol/l磷酸盐缓冲溶液(ph 7.5)适当稀释后,测定在不同温度下(25℃~90℃)儿茶酚1,2-双加氧酶的活性,以儿茶酚1,2-双加氧酶活性最高者为100%。
98.2、儿茶酚1,2-双加氧酶的热稳定性
99.将实施例4制备的儿茶酚1,2-双加氧酶粗酶液用50mmol/l磷酸盐缓冲溶液(ph 7.5)适当稀释后,测定在不同温度下保温不同时间后残存的儿茶酚1,2-双加氧酶活力,以初始儿茶酚1,2-双加氧酶活力为100%。
100.实验结果表明,儿茶酚1,2-双加氧酶的最适反应温度为65℃,儿茶酚1,2-双加氧酶在25℃~90℃范围内能稳定超过8h,实验结果如图6所示。
101.2、儿茶酚1,2-双加氧酶的最适ph及ph耐受性研究
102.1)儿茶酚1,2-双加氧酶的最适ph
103.将实施例4制备的儿茶酚1,2-双加氧酶粗酶液用50mmol/l磷酸盐缓冲溶液(ph 7.5)适当稀释后,测定儿茶酚1,2-双加氧酶的活性,以儿茶酚1,2-双加氧酶活性最高者为
100%。
104.2)儿茶酚1,2-双加氧酶的ph稳定性
105.将实施例4制备的儿茶酚1,2-双加氧酶粗酶液用50mmol/l磷酸盐缓冲溶液(ph 7.5)适当稀释后,最适温度下保温3h,测定残存的酶活性,以初始酶活力为100%。
106.实验结果表明,儿茶酚1,2-双加氧酶的最适反应ph为7.0,ph在2.0~8.0的范围内,保存3h仍具有高于45%的活性,实验结果如图7所示。
107.实施例7
108.该实施例提供了一种儿茶酚1,2-双加氧酶对重油微生物降解的影响分析方法,具体包括:
109.(1)菌群的重油降解
110.将重油降解菌群接种于30ml含有0.01g重油的无机培养基中,以不接种菌悬液的实验组作为空白对照,30℃、160r/min恒温摇床震荡培养12天。分别取第0、2、4、6、8、10、12、14天的完整培养液测定重油浓度的变化,考察菌群的重油降解效率。每个实验设置四组平行。
111.(2)儿茶酚1,2-双加氧酶-菌群的重油降解
112.接种已优化添加量的儿茶酚1,2-双加氧酶粗酶液(3.03u/ml)于含有重油降解菌群的30ml含有0.01g重油的无机培养基中,以不接种儿茶酚1,2-双加氧酶-菌群的实验组作为空白对照,30℃、160r/min恒温摇床震荡培养12天。分别取第0、2、4、6、8、10、12、14天的完整培养液测定重油浓度的变化,考察儿茶酚1,2-双加氧酶-菌群的重油降解效率。每个实验设置四组平行。
113.(3)重油降解效率和重油降解动力学分析
114.采用红外分光光度法测定培养液中残余的重油浓度。第n日重油的降解效率按照下式计算。
[0115][0116]
式中,c0为底物的原始浓度,cn为第n天的底物浓度,单位均为(mg/l)。
[0117]
重油的微生物降解过程一般符合一级反应动力学,可按照下述反应方程进行拟合,以求得重油微生物降解的反应速率k(d-1
)。
[0118]
lnc=-kt+b。
[0119]
式中,c为t时的重油浓度(mg/l),t为重油降解时间(d)。
[0120]
在降解10天后,添加菌群和儿茶酚1,2-双加氧酶-菌群联合作用体系的培养基中重油浓度分别从223.59mg/l下降到88.10mg/l和88.27mg/l,菌群和儿茶酚1,2-双加氧酶-菌群联合作用体系的重油降解效率分别为60.60%和60.52%;重油的生物降解速率常数从0.1038d-1
上升到0.1586d-1
;尽管儿茶酚1,2-双加氧酶的添加不能促进微生物的底物作用范围,但能提高重油的生物降解效率,实验结果如图8~10所示。
[0121]
需要说明的是,上述实施例所使用的材料如下:
[0122]
1、菌株和质粒
[0123]
节杆菌arthrobacter sp.fb24购自中国工业微生物菌种保藏管理中心(cic c)。
克隆用菌株e.coli dh5α,表达菌株e.coli bl21(de3)为实验室储藏菌株,质粒pmd19t,pet28a为实验室储藏质粒。
[0124]
2、酶、生化试剂和底物
[0125]
dna聚合酶和所有的限制性内切酶购自takara(japan)公司。iptg、卡那霉素、咪唑购自sigma(usa)公司。dna提取试剂盒、ni-nta agarose纯化柱购自qiagen(germany)公司。双预染蛋白marker购自tiangen(china)生化有限公司。其他试剂均为分析纯,购自北京江晨生物科技有限公司。
[0126]
3、培养基
[0127]
1)lb培养基(g/l)
[0128]
nacl 10g,胰蛋白胨10g,酵母提取物5g,蒸馏水定容至1l,ph 7.2~7.4;固体培养基添加20g琼脂粉;121℃高温高压蒸汽灭菌30min后备用。
[0129]
2)lb-amp培养基
[0130]
100ml lb培养基中加入50μl氨苄青霉素(100mg/ml,ampicillin,a mp)使amp的终浓度为50μg/ml,即配得lb-amp培养基。
[0131]
lb-amp固体培养基加入2%的琼脂即得。
[0132]
3)lb-kan培养基
[0133]
100ml lb培养基中加入50μl卡那霉素(100mg/ml,kanamycin,kan)使kan的终浓度为50μg/ml,即配得lb-kan培养基。
[0134]
4)无机盐培养基(g/l)
[0135]
将na2hpo4·
7h2o 7g,kh2po43.0 g,(nh4)2so45 g,nacl 3g,mg so4·
7h2o 0.7g混合,并用蒸馏水定容至1l,ph 7.5;固体培养基添加20g琼脂粉;于121℃高压蒸汽灭菌30min后备用。
[0136]
5)重油培养基
[0137]
无机盐培养基中加入一定量的重油,于121℃高压蒸汽灭菌30min后备用。重油组分分布为烷烃36%、芳烃35%、胶质20%及沥青质9%。
[0138]
4、琼脂糖凝胶电泳所需溶液
[0139]
1)50
×
tae(tris-乙酸)缓冲液
[0140]
称取tris碱242g,na2edta.2h2o 37.2g,加入900ml的ddh2o,充分搅拌溶解。加入57.1ml的醋酸,充分混匀,加入ddh2o定容至1l,室温保存。
[0141]
在进行琼脂糖凝胶电泳时,需使用1
×
tae缓冲溶液,此时取20ml 50
×
ta e缓冲溶液定容至1l即可。
[0142]
2)溴化乙锭(eb)
[0143]
称取10mg溴化乙锭,加入ddh2o定容至100ml,于黑暗处静置保存。
[0144]
5、十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sds-page)所需溶液
[0145]
1)500ml 1mol/l tris-hcl(ph6.8)
[0146]
称取60.57gtris碱溶于300ml的去离子水,充分搅拌溶解,加入0.1mol/l的hcl调节到ph至6.8(ph至7.10需要45.7ml 0.1mol/l盐酸),加水调节至500ml,室温保存。
[0147]
2)浓缩buffer(上层胶buffer)
[0148]
250ml浓缩胶buffer:取250ml 1mol/l tris-hcl加入25ml 10%sds
[0149]
(终浓度1%),混匀。
[0150]
3)500ml 1.5mol/l tris-hcl(ph8.8)
[0151]
称取90.86g tris碱溶于300ml的去离子水,搅拌至充分溶解,加入0.1mol/l hcl(约8.5ml hcl)调节所需要的ph至8.8,将溶液定容至500ml,室温保存。
[0152]
4)分离buffer(下层胶buffer)
[0153]
250ml分离胶buffer:取250ml 1.5mol/l tris-hcl加入25ml 10%s ds(终浓度1%),混匀。
[0154]
5)考马斯亮蓝r-250染色液
[0155]
1l甲醇:乙酸溶液:
[0156]
将900ml甲醇:水(450ml甲醇和450ml水)与100ml冰醋酸混合。
[0157]
在每500ml甲醇:乙酸溶液中溶解1.25g考马斯亮蓝r-250,用whatman i号滤纸过滤去除颗粒物。
[0158]
6)脱色液(1l)
[0159]
甲醇:乙酸溶液即为洗脱液。
[0160]
将900ml甲醇:水(450ml甲醇和450ml水)与100ml冰醋酸混合
[0161]
7)10%过硫酸铵(10%aps)
[0162]
在1ml水中溶解0.1g过硫酸铵。
[0163]
8)5
×
sds-page buffer(分子克隆)
[0164]
称取15.1gtris,94g甘氨酸(glycine),50ml 10%sds(终浓度0.5%),用500ml去离子水溶解,搅拌至充分溶解,定容至1000ml,室温保存。
[0165]
9)5
×
sds-page加样缓冲液(loading buffer)
[0166]
分别取1.25ml 1mol/l tris-hcl(ph6.8),0.5g sds,0.025g溴酚蓝,2.5ml甘油,于10ml离心管中,加去离子水溶解后,定容至5ml,小份(0.5ml/份)分装,于室温保存。使用前将18μl(17.86μl)14mol/l的2-me加入到每小份中,在室温下可保存一个月。
[0167]
10)10%sds
[0168]
10g sds溶于100ml蒸馏水中。
[0169]
11)30%丙烯酰胺:双甲叉丙烯酰胺(29:1)
[0170]
0.5g双甲叉丙烯酰胺溶解在30ml水中,然后加入14.5g丙烯酰胺,溶解后用水定容至50ml。
[0171]
6、ni-nta填料纯化蛋白缓冲溶液
[0172]
1)裂解缓冲溶液(1l)
[0173]
50mmol/l nah2po4(6.0g nah2po4),300mmol/l nacl(17.54g na cl),10mmol/l咪唑(0.68g咪唑),加入ddh2o 900ml,利用naoh调节ph至8.0,加ddh2o定容至1l。
[0174]
2)洗脱缓冲溶液(wash buffer,1l)
[0175]
50mmol/l nah2po4(6.0g nah2po4),300mmol/l nacl(17.54g nac l),20mmol/l(1.36g咪唑),加入ddh2o 900ml,利用naoh调节ph至8.0,加ddh2o定容至1l。
[0176]
3)eb缓冲溶液(elution buffer,1l)
[0177]
50mmol/l nah2po4(6.0g nah2po4),300mmol/l nacl(17.54g na cl),250mmol/l咪唑(17.00g咪唑),加入ddh2o 900ml,利用naoh调节ph至8.0,加ddh2o定容至1l。
[0178]
7、凝胶的配置
[0179]
1)琼脂糖凝胶的配置
[0180]
于100ml 1
×
tae缓冲液中加入0.7g琼脂糖,微波炉中加热至完全溶解。
[0181]
2)sds-page凝胶的配置
[0182]
sds-page凝胶包括上层的浓缩胶和下层的分离胶,具体的配置方法如表1所示。
[0183]
表1分离胶与浓缩胶的配制
[0184][0185][0186]
另外,质粒的提取的方法如下:
[0187]
1)挑取含质粒的单菌落,接种到3ml含适当抗生素的液体lb培养基中,于恒温摇床37℃、160r/min振荡培养过夜;
[0188]
2)将过夜培养的菌液分装于1.5ml离心管中,室温12000r/min离心1m in收集菌体,弃上清;
[0189]
3)取100μl预冷的溶液i加入每个离心管中,混合均匀后,冰浴3分钟;
[0190]
4)每管各加入新配置的溶液ii 200μl,颠倒混匀,冰浴3min;
[0191]
5)每管各加入150μl溶液iii,颠倒混匀,冰浴3min;
[0192]
6)上层水相用等体积的酚:氯仿(500μl),12000r/min离心7min,取上清;
[0193]
7)12000r/min离心10min,沉淀用70%乙醇洗一遍,离心干燥;
[0194]
8)将沉淀物溶于20μl的无菌水中,加入适量的rnasea(10mg/ml),于37℃保温15~30min,质粒提取成功。将含有质粒的溶液于-20℃保存。
[0195]
其中溶液i、ii和iii的组成分别如下:
[0196]
溶液i:25mmol/l tris-hcl(ph 7.4),10mmol/l edta(ph 8.0),50mmol/l葡萄糖,高压灭菌,4℃保存。
[0197]
溶液ii:1mol/l naoh,1%sds,以1:1比例混合。配置时各稀释10倍。
[0198]
溶液iii:3mol/l kac(ph 4.8)。
[0199]
电转感受态细胞的制备方法如下:
[0200]
1)取新鲜的e.coli dh5α或e.coli bl21(de3)菌落接种于10ml lb液体培养基中,于恒温摇床37℃、160r/min振荡培养过夜;
[0201]
2)次日按1%接种量接种于新鲜的lb液体培养基中,于37℃,300r/min振荡培养3
~5h,使od
600
达到0.5~0.8;
[0202]
3)将培养物于冰水混合物中冰浴15~30min,4℃、4000r/min离心10min,尽可能地去掉上清;
[0203]
4)将培养物于冰水混合物中冰浴15~30min,4℃、4000r/min离心10min,去掉上清;
[0204]
5)加入500ml预冷灭菌的超纯水,轻轻翻转重悬菌体,按照(4)的方法离心去上清;再次用300ml预冷灭菌的超纯水悬浮菌体,再次按照(4)的方法离心去除上清;
[0205]
6)取100ml预冷的10%甘油清洗菌体1次;
[0206]
7)用大约1ml预冷的10%甘油悬浮菌体;
[0207]
8)按40μl每份分装细胞悬液后置于-80℃冰箱保存备用。
[0208]
电转化的方法如下:
[0209]
1)将e.coli dh5α或e.coli bl21(de3)电转感受态细胞置于冰上溶解,将电转化杯和白色滑块放在0℃预冷;
[0210]
2)调节bio-rad电转化仪的参数:pulser apparatus 25μf,pulser control ler 200ω;0.2cm电激槽用2.5kv,0.1cm电激槽用1.5~1.8kv;
[0211]
3)取1~2μl连接产物于40μl感受态细胞中,混合均匀,冰上放置1~2min;
[0212]
4)将混合物吸入预冷的电激杯内,轻甩使溶液置于底部,迅速将电激杯放入预冷的滑块内,并将其推入,同时按下两个pulse键;
[0213]
5)迅速取出电激槽,立即加入960μl lb液体培养基,混匀,移取至1.5ml离心管中,37℃、250r/min震动培养0.5~1h;
[0214]
6)取100μl在lb-amp或lb-kan培养基上涂布培养,筛选转化子。
[0215]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1