一种吲唑酰肼类化合物及其应用
本发明属于药物合成技术领域,尤其涉及一种吲唑酰肼类化合物及其应用。
背景技术:
前列腺癌是我国与西方国家男性中最常见的恶性肿瘤之一,并且随着我国人口不断老龄化以及生活习惯的改变,前列腺癌的发病率快速上升,并且三分之二的前列腺癌患者在确诊时已是晚期,失去根治性治疗的机会。以抗雄激素药物为主的内分泌治疗是晚期前列腺癌的首选治疗方式,初期能获得满意疗效,但12至18个月后,大部分患者会逐渐发展为具有高转移性的去势抵抗性前列腺癌(castrationresistantprostatecancer,crpc),其中90%转移性crpc发生在骨骼。
针对第一代抗雄激素药物产生耐药性的crpc患者,临床推荐使用阿比特龙、恩杂鲁胺等新型抗雄激素药物。尽管阿比特龙、恩杂鲁胺能够延长crpc患者的生存期,但是临床实践和大型临床试验研究表明新型抗雄激素药物也无法避免原发性或获得性耐药的产生。
细胞外基质与肿瘤细胞相互作用是crpc耐药性形成的驱动因素。肿瘤细胞与细胞外基质(extracellularmatrix,ecm)之间的相互作用促进肿瘤细胞增殖、侵袭及转移。近来研究表明,ecm介导的耐药性是影响肿瘤进展和治疗反应的重要因素。在骨转移crpc中,许多整合素家族,tgf-β家族,骨驻留蛋白,rankl和pthrp等参与基质重构,影响前列腺癌的耐药性。因此,开发靶向肿瘤细胞和微环境基质相互作用的药物有望成为对抗crpc耐药性的新治疗策略。
整合素受体是影响肿瘤转移及耐药性的重要细胞外基质。其中,αvβ3受体在肿瘤细胞和宿主基质细胞上表达,阻断细胞外基质与整合素受体αvβ3的相互作用,可以发挥抗肿瘤及转移的作用。西仑吉肽是avβ3的整合素受体抑制剂,在不同类型肿瘤中开展了多项ii期临床试验,其中包括去势耐药的前列腺癌。虽然,西仑吉肽单药治疗有很好的耐受性,但是抗肿瘤活性相对较弱,最终西仑吉肽3期临床试验以失败告终。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种对整合素avβ3受体具有较好抑制作用的吲唑酰肼类化合物及其应用。
本发明提供了一种吲唑酰肼类化合物,如式(i)所示:
其中,r选自取代的烷基、取代的烯基或取代的苯基;
所述取代的烷基与取代的烯基中的取代基包括苯基和/或取代的苯基;
r′选自h或烷基。
优选的,所述r选自取代的c2~c10的烷基、取代的c2~c10的烯基或取代的苯基;
所述r′选自h或c1~c10的烷基。
优选的,所述取代的苯基中的取代基选自c1~c5的烷基、c1~c5的烯基、羟基、c1~c5的烷氧基、硝基与c1~c5的卤代烷基中的一种或多种。
优选的,所述吲唑酰肼类化合物选自式(i-1)~式(i-5)中的一种:
其中,n与m各自独立地为1~5的整数;
r1~r8各自独立地选自c1~c5的烷基、c1~c5的烯基、羟基、c1~c5的烷氧基、硝基或c1~c5的卤代烷基;
所述r′选自h或c1~c5的烷基。
优选的,所述n与m各自独立地为1~3的整数;
r1~r8各自独立地选自c1~c3的烷基、c1~c3的烯基、羟基、c1~c3的烷氧基、硝基或c1~c3的卤代烷基;
所述r′选自h或c1~c3的烷基。
优选的,所述吲唑酰肼类化合物选自式(1)~式(12)中的一种:
本发明还提供了上述吲唑酰肼类化合物作为整合素avβ3受体拮抗剂的应用。
本发明还提供了上述吲唑酰肼类化合物在制备肿瘤药物中的应用。
优选的,所述肿瘤为前列腺癌、黑色素瘤与卵巢癌中的一种或多种。
优选的,所述肿瘤为对恩杂鲁胺治疗具有耐药性的肿瘤。
本发明提供了一种吲唑酰肼类化合物,如式(i)所示;其中,r选自取代的烷基、取代的烯基或取代的苯基;所述取代的烷基与取代的烯基中的取代基包括苯基和/或取代的苯基;r′选自h或烷基。与现有技术相比,本发明提供的吲哚酰肼类化合物可作为整合素avβ3受体拮抗剂,同时具有明显的抗前列腺癌活性,并且针对恩杂鲁胺耐药的细胞系具有显著性抑制作用。
实验表明,式(5)所示的吲唑酰肼类化合物与蛋白αvβ3相互作用的kd值为158nm;式(5)所示的吲唑酰肼类化合物对多种肿瘤细胞有效,包括前列腺癌、黑色素瘤、卵巢癌等,并对血管内皮细胞也具有抑制作用。并针对前列腺癌恩杂鲁胺耐药的22rv1细胞进行联合用药研究,发现式(5)所示的吲唑酰肼类化合物与恩杂鲁胺有协同作用。
附图说明
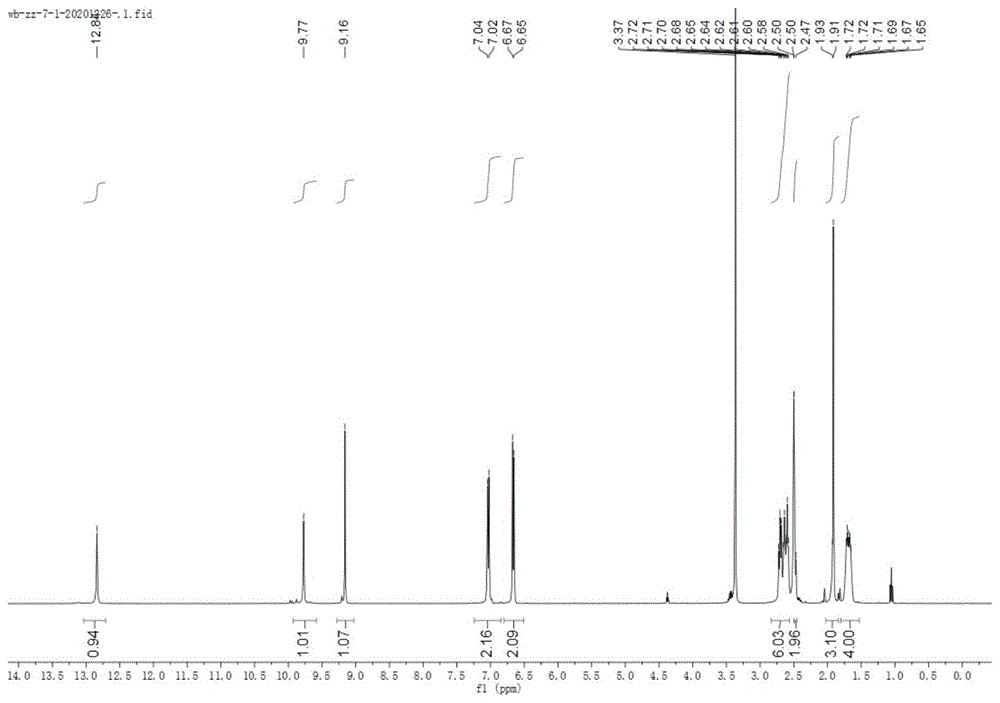
图1为本发明实施例1中式(1)所示的化合物的核磁共振氢谱图;
图2为本发明实施例1中式(1)所示的化合物的核磁共振碳谱图;
图3为本发明实施例1中式(3)所示的化合物的核磁共振氢谱图;
图4为本发明实施例1中式(3)所示的化合物的核磁共振碳谱图;
图5为本发明实施例1中式(4)所示的化合物的核磁共振氢谱图;
图6为本发明实施例1中式(4)所示的化合物的核磁共振碳谱图;
图7为本发明实施例1中式(5)所示的化合物的核磁共振氢谱图;
图8为本发明实施例1中式(5)所示的化合物的核磁共振碳谱图;
图9为本发明实施例1中式(6)所示的化合物的核磁共振氢谱图;
图10为本发明实施例1中式(6)所示的化合物的核磁共振碳谱图;
图11为本发明实施例1中式(7)所示的化合物的核磁共振氢谱图;
图12为本发明实施例1中式(7)所示的化合物的核磁共振碳谱图;
图13为本发明实施例1中式(8)所示的化合物的核磁共振氢谱图;
图14为本发明实施例1中式(8)所示的化合物的核磁共振碳谱图;
图15为本发明实施例1中式(9)所示的化合物的核磁共振氢谱图;
图16为本发明实施例1中式(9)所示的化合物的核磁共振碳谱图;
图17为本发明实施例1中式(10)所示的化合物的核磁共振氢谱图;
图18为本发明实施例1中式(10)所示的化合物的核磁共振碳谱图;
图19为本发明实施例2中吲唑酰肼类化合物配制和稀释示意图;
图20为本发明实施例2中待测药物加样示意图;
图21为本发明实施例2中式(5)所示的化合物对不同肿瘤细胞的活性曲线图;
图22为本发明实施例2中式(5)所示的化合物对不同肿瘤细胞的活性曲线图;
图23为利用化学发光测定式(5)所示的化合物与恩杂鲁胺针对细胞22rv1联合用药得到的拟合药效计量曲线图;
图24为式(1)所示的化合物(代号19-1)、式(3)所示化合物(代号19-2)、式(2)所示的化合物(代号19-3)、式(4)所示的化合物(代号19-4)与式(10)所示的化合物(代号19-5)针对肿瘤细胞22rv1的活性曲线图;
图25为式(7)所示化合物(代号19-6)、式(8)所示化合物(代号19-7)、式(6)所示化合物(代号19-8)与式(9)所示化合物(代号19-9)针对肿瘤细胞22rv1的活性曲线图;
图26为式(1)所示的化合物(代号19-1)、式(3)所示化合物(代号19-2)、式(2)所示的化合物(代号19-3)、式(4)所示的化合物(代号19-4)与式(10)所示的化合物(代号19-5)针对肿瘤细胞lncap的活性曲线图;
图27为式(7)所示化合物(代号19-6)、式(8)所示化合物(代号19-7)、式(6)所示化合物(代号19-8)与式(9)所示化合物(代号19-9)针对肿瘤细胞lncap的活性曲线图。
具体实施方式
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供了一种吲唑酰肼类化合物,如式(i)所示:
其中,r选自取代的烷基、取代的烯基或取代的苯基,优选为取代的c2~c10的烷基、取代的c2~c10的烯基或取代的苯基,更优选为取代的c2~c8的烷基、取代的c2~c8的烯基或取代的苯基,再优选为取代的c2~c6的烷基、取代的c2~c6的烯基或取代的苯基;所述取代的烷基与取代的烯基中的取代基包括苯基和/或取代的苯基。
r′为h或烷基,优选为h或c1~c10的烷基,更优选为h或c1~c5的烷基,再优选为h或c1~c3的烷基,最优选为h或甲基。
按照本发明,所述取代的苯基中的取代基优选为c1~c5的烷基、c1~c5的烯基、羟基、c1~c5的烷氧基、硝基与c1~c5的卤代烷基中的一种或多种,更优选为c1~c3的烷基、c1~c3的烯基、羟基、c1~c3的烷氧基、硝基与c1~c3的卤代烷基中的一种或多种,再优选为c1~c2的烷基、c1~c2的烯基、羟基、c1~c2的烷氧基、硝基与c1~c2的卤代烷基中的一种或多种;所述卤代烷基中的卤原子优选为氟、氯、溴或碘,更优选为氟;所述卤代烷基中卤原子的数量优选为1~3。
在本发明中,进一步具体地,所述吲唑酰肼类化合物选自式(i-1)~式(i-5)中的一种:
其中,n与m各自独立地为1~5的整数,更优选为1~4的整数,再优选为1~3的整数,最优选为1或2。
r1~r8各自独立地优选为c1~c5的烷基、c1~c5的烯基、羟基、c1~c5的烷氧基、硝基或c1~c5的卤代烷基,更优选为c1~c3的烷基、c1~c3的烯基、羟基、c1~c3的烷氧基、硝基与c1~c3的卤代烷基中的一种或多种,再优选为c1~c2的烷基、c1~c2的烯基、羟基、c1~c2的烷氧基、硝基与c1~c2的卤代烷基中的一种或多种;所述卤代烷基中的卤原子优选为氟、氯、溴或碘,更优选为氟;所述卤代烷基中卤原子的数量优选为1~3。
最优选地,所述吲唑酰肼类化合物选自式(1)~式(12)中的一种:
本发明提供的吲哚酰肼类化合物可作为整合素avβ3受体拮抗剂,同时具有明显的抗前列腺癌活性,并且针对恩杂鲁胺耐药的细胞系具有显著性抑制作用。
本发明还提供了一种上述吲唑酰肼类化合物的制备方法,包括:将式(ii)所示的化合物与rcor′反应,得到式(i)所示的化合物。
所述式(ii)所示的化合物与rcor′优选在溶剂中反应;所述溶剂优选为醇溶剂,更优选为乙醇;所述反应优选为回流反应;所述式(ii)所示的化合物与rcor′的摩尔比优选为1:(1~1.2);在本发明提供的实施例中具体为1:1.1。
所述式(ii)所示的化合物优选按照以下步骤进行:将式(iii)所示的化合物与联氨反应,得到式(ii)所示的化合物。
其中,r〃为c1~c5的烷基,更优选为c1~c4的烷基,再优选为乙基。
所述式(iii)所示的化合物与联氨优选在溶剂中反应;所述溶剂优选为醇溶剂,更优选为乙醇;式(iii)所示的化合物与联氨的摩尔比优选为1:(6~8),更优选为1:7;所述反应优选为回流反应;所述反应的时间优选为20~30h;反应结束后除去溶剂,用乙酸乙酯与水洗涤,得到式(ii)所示的化合物。
所述式(iii)所示的化合物优选按照以下步骤进行制备:将式(iv)所示的化合物与联氨在乙酸中加热反应,得到式(iii)所示的化合物。
所述式(iv)所示的化合物与联氨的摩尔比优选为1:(1~1.2),更优选为1:(1.1~1.2);所述加热反应优选为回流反应;所述加热反应的时间优选为40~70min,更优选为60min;加热反应结束后,优选倒入冰水中,用碳酸氢钠中和后用乙酸乙酯萃取,萃取液浓缩后用正己烷与乙酸乙酯的混合液进行柱层析纯化,得到式(iii)所示的化合物;所述正己烷与乙酸乙酯的体积比优选为3:1。
所述式(iv)所示的化合物优选按照以下步骤进行制备:将环己酮与(coor′)在乙醇钠溶液中反应,然后硫酸溶液分解反应混合物后,用乙酸乙酯萃取,萃取液浓缩后用正己烷与乙酸乙酯的混合液柱层析纯化,得到式(iv)所示的化合物;所述反应的时间优选为10~14h,更优选为12h;所述正己烷与乙酸乙酯的体积优选为12:1。
本发明还提供了上述吲唑酰肼类化合物作为整合素avβ3受体拮抗剂的应用。
本发明还提供了上述吲唑酰肼类化合物在制备肿瘤药物中的应用。
其中,所述肿瘤优选为前列腺癌、黑色素瘤与卵巢癌中的一种或多种。
进一步具体地,所述肿瘤优选为对恩杂鲁胺治疗具有耐药性的肿瘤。
进一步具体地,本发明提供了上述吲唑酰肼类化合物制备抑制肿瘤细胞22rv1、22rv1-spp1及lncap药物中的应用。
为了进一步说明本发明,以下结合实施例对本发明提供的一种吲唑酰肼类化合物及其应用进行详细描述。
以下实施例中所用的试剂均为市售。
实施例1
按照以下步骤制备式(1)、式(2)~式(10)所示的化合物:
将钠(1.5g,65mmol)添加到0℃的无水乙醇(20ml)中制备乙醇钠溶液;然后缓慢添加环己酮(4.41g,44mmol)和草酸二乙酯(7.3g,50mmol)的混合物,并在室温下搅拌该溶液12h,再用2n硫酸溶液分解反应混合物后,用乙酸乙酯萃取混合物,干燥并浓缩有机溶剂,得到的粗产物用正己烷:乙酸乙酯(12:1)柱层析进一步纯化,得到黄色油即为产物2(5.87g,67%)。
将联氨(448mg,14mmol)缓慢地添加到产物2(2.38g,12mmol)的乙酸(5ml)冷却悬浮液中,将混合物加热回流1h,倒入冰水中,用nahco3中和,并用乙酸乙酯萃取,合并有机相,干燥(na2so4)、过滤并浓缩,再用正己烷:乙酸乙酯(3:1)柱层析纯化残余物,得到白色固体即为产物3(2g,86%)。
将产物3(970mg,5mmol)和联氨(1.34g,35mmol)在乙醇(10ml)中加热至回流1天。回流结束后,蒸发乙醇并通过过滤收集沉淀物,用乙酸乙酯和水洗涤,得到白色固体形式的酰肼4(580mg,64%)。
将酰肼4(72mg,0.4mmol)和取代醛rcor′(0.44mmol)在乙醇(3ml)中回流以获得沉淀物化合物5,然后通过过滤收集沉淀物并用冷乙醇(81%)洗涤。
利用核磁共振对得到的式(1)、式(3)~式(10)所示的化合物进行分析,得到谱图如图1~图18所示;其中图1为式(1)所示的化合物的核磁共振氢谱图;图2为式(1)所示的化合物的核磁共振碳谱图;图3为式(3)所示的化合物的核磁共振氢谱图;图4为式(3)所示的化合物的核磁共振碳谱图;图5为式(4)所示的化合物的核磁共振氢谱图;图6为式(4)所示的化合物的核磁共振碳谱图;图7为式(5)所示的化合物的核磁共振氢谱图;图8为式(5)所示的化合物的核磁共振碳谱图;图9为式(6)所示的化合物的核磁共振氢谱图;图10为式(6)所示的化合物的核磁共振碳谱图;图11为式(7)所示的化合物的核磁共振氢谱图;图12为式(7)所示的化合物的核磁共振碳谱图;图13为式(8)所示的化合物的核磁共振氢谱图;图14为式(8)所示的化合物的核磁共振碳谱图;图15为式(9)所示的化合物的核磁共振氢谱图;图16为式(9)所示的化合物的核磁共振碳谱图;图17为式(10)所示的化合物的核磁共振氢谱图;图18为式(10)所示的化合物的核磁共振碳谱图。
得到核磁共振结果如下:
式(1)所示的吲唑酰肼类化合物:
1hnmr(400mhz,dmso-d6)δ12.84(s,1h),9.77(s,1h),9.16(s,1h),7.03(d,j=8.3hz,2h),6.66(d,j=8.4hz,2h),2.75–2.56(m,6h),2.50–2.45(m,2h),1.91(s,3h),1.76–1.60(m,4h).
13cnmr(101mhz,dmso-d6)δ158.54,157.05,155.46,141.29,140.16,131.42,129.22,116.34,115.08,40.77,31.28,22.62,21.98,20.97,20.60,15.55.
式(3)所示的吲唑酰肼类化合物:
1hnmr(400mhz,dmso-d6)δ12.86(s,1h),11.16(s,1h),8.31(s,1h),7.43–7.28(m,3h),7.17–7.12(m,2h),6.07(t,j=7.6hz,1h),2.57(d,j=6.0hz,4h),1.95(t,j=7.2hz,2h),1.76–1.58(m,5h),0.82(d,j=6.6hz,6h).
13cnmr(101mhz,dmso-d6)δ158.89,151.63,141.23,139.95,139.65,139.57,135.93,129.64,127.86,127.03,116.55,37.79,28.13,22.60,22.33,21.96,20.93,20.56.
式(4)所示的吲唑酰肼类化合物:
1hnmr(400mhz,dmso-d6)δ12.89(s,1h),11.41(s,1h),8.45(s,1h),7.55(d,j=7.8hz,2h),7.25(d,j=7.8hz,2h),2.65(dt,j=25.1,6.2hz,4h),2.33(s,3h),1.78–1.62(m,4h).
13cnmr(101mhz,dmso-d6)δ159.10,146.65,141.19,139.95,139.39,131.99,129.34,126.79,116.62,22.58,21.94,20.96,20.96,20.54.
式(5)所示的吲唑酰肼类化合物:
1hnmr(400mhz,dmso-d6)δ12.92(s,1h),11.46(s,1h),9.62(s,1h),8.38(s,1h),7.23(t,j=7.8hz,1h),7.13(s,1h),7.03(d,j=7.5hz,1h),6.80(dd,j=8.1,2.4hz,1h),2.64(dt,j=21.7,6.1hz,4h),1.71(dd,j=13.7,6.6hz,4h).
13cnmr(101mhz,dmso-d6)δ159.17,157.65,146.77,141.18,140.06,136.01,129.86,118.55,117.09,116.73,112.50,22.63,21.99,21.05,20.59.
式(6)所示的吲唑酰肼类化合物:
1hnmr(400mhz,dmso-d6)δ13.00(s,1h),11.89(s,1h),8.59(s,1h),8.29(d,j=8.6hz,2h),7.91(d,j=8.5hz,2h),2.65(dt,j=22.2,6.2hz,4h),1.70(dd,j=13.7,6.6hz,4h).
13cnmr(101mhz,dmso-d6)δ159.41,147.61,144.16,141.14,140.92,140.23,127.74,124.14,117.05,22.60,21.97,21.03,20.58.
式(7)所示的吲唑酰肼类化合物:
1hnmr(400mhz,dmso-d6)δ12.91(s,1h),11.39(s,1h),9.57(s,1h),8.35(s,1h),7.20(s,1h),7.11(d,j=7.7hz,1h),6.92(d,j=7.5hz,1h),2.64(dt,j=22.6,6.2hz,4h),2.13(s,3h),1.71(dd,j=13.4,6.8hz,4h).
13cnmr(101mhz,dmso-d6)δ159.13,155.70,146.97,141.27,140.05,133.47,130.85,126.44,118.89,116.68,111.42,22.66,22.02,21.08,20.62,16.11.
式(8)所示的吲唑酰肼类化合物:
1hnmr(400mhz,dmso-d6)δ12.91(s,1h),11.41(s,1h),8.39(s,1h),7.29(s,1h),7.11(d,j=8.2hz,1h),7.01(d,j=8.3hz,1h),3.81(s,3h),3.79(s,3h),2.64(dt,j=23.9,6.2hz,4h),1.71(dq,j=12.6,6.5,5.0hz,4h).
13cnmr(101mhz,dmso-d6)δ159.13,150.48,149.05,146.83,141.29,140.05,127.47,121.61,116.68,111.45,107.96,55.57,55.41,22.66,22.02,21.05,20.62.
式(9)所示的吲唑酰肼类化合物:
1hnmr(400mhz,dmso-d6)δ12.88(s,1h),11.30(s,1h),9.28(s,1h),8.32(s,1h),7.21(s,1h),7.02–6.88(m,2h),3.79(s,3h),2.70–2.56(m,4h),1.80–1.61(m,4h).
13cnmr(101mhz,dmso-d6)δ159.02,149.49,146.85,146.82,141.30,139.97,127.57,119.91,116.58,112.26,111.86,55.56,22.64,21.99,21.04,20.59.
式(10)所示的吲唑酰肼类化合物:
1hnmr(400mhz,dmso-d6)δ12.91(s,1h),11.72(s,1h),11.10(s,2h),9.74(s,1h),8.80(s,1h),5.81(s,2h),2.63(dt,j=21.7,6.2hz,4h),1.85–1.52(m,4h).
13cnmr(101mhz,dmso-d6)δ161.11,159.60,158.67,145.98,140.93,139.99,116.70,99.36,94.30,56.08,22.67,22.02,21.01,20.60,18.62.
实施例3:抗肿瘤活性检测
3.1实验方法
3.1.1肿瘤细胞复苏培养
从液氮中取出冻存的肿瘤细胞(前列腺癌、卵巢癌等细胞),在37℃快速溶解,加入5ml含有fbs的对应培养基中,300g离心5分钟,收集沉淀,用对应培养基重悬后,充分混合后,加入24孔细胞培养皿中,补充培养基到每孔总体积2ml37℃培养到汇合度约90%,传代扩增或计数铺板。
3.1.2肿瘤细胞接种
待肿瘤细胞培养到可以传代,用胰酶消化并制备单细胞悬液。细胞计数后,用对应培养基调整细胞浓度到1.2×104cells/ml,在96孔板中加入90μl混合液,置于37℃孵育30分钟,加入对应培养,在37℃细胞培养箱中培养过夜,观察肿瘤细胞的形态和汇合度。
3.1.3化合物配制和稀释
当肿瘤细胞过夜培养后,制备化合物梯度稀释液并加入培养体系。
将吲唑酰肼类化合物溶解于dmso中的储液(100mm)在室温充分溶解,如果有不溶物,超声加热5分钟再观察,直至溶解。
在96孔v形化合物稀释板中的2-9号孔分别加入20μldmso,在1孔中加入30μl储液,将化合物稀释板至于震荡混匀器上,从1号孔转移10μl液体加入2号孔,吹打震荡混匀后,从2号孔转移10μl液体到3号孔,震荡吹大混匀,依次类推…直至从8号孔转移10μl液体到9号孔,吹打震荡混匀,制备1000×梯度稀释储液完成。【注意:dmso容易吸收空气中的水分,操作完后,立即加封口膜封闭化合物稀释板,储存于4摄氏度,1周后丢弃。再次使用时,温度达到室温后再开封口膜,避免吸湿造成浓度不准确。】
取96孔无菌细胞培养板,于超净工作台中,在1-9号孔加入198μl培养基,用微量加样器从1000×储液中,对应1-9号孔转移2μl储液到96孔无菌细胞培养板对应的1-9号孔,震荡吹打混匀,制备10×储液完成。本储液需要当天配制使用。
在细胞培养基中加入10μl当天制备好的10×储液。
参见图19,图19为吲唑酰肼类化合物配制和稀释示意图。
3.1.4加待测药物
待观察肿瘤细胞贴壁生长良好后,对应platemap依此加入10μl相应浓度的化合物,当天制备好的10×储液。置于37℃,5%二氧化碳条件下孵育72h。
参见图20,图20待测药物加样示意图,其中,null:空白孔;dmso:加入dmso的孔;bez235:2.5μm的bez235;staurosporine:1μmstaurosporine;cpd#19为式(5)所示的酰胺类化合物。
3.1.5化学发光测定
采用化学发光法测定细胞atp水平,进而评价细胞活力。具体操作按照说明书操作,培养结束后加入50μlctg溶液,混匀后,裂解混合物转移到酶标板中,5~10分钟后,在酶标仪中采集化学发光数据。使用excel软件分析处理数据,使用graphpadprism7软件,根据化学发光数据的拟合药效计量曲线,计算ic50。
3.1.6对照和质控
本试验用zfactor作为质控指标。z’factor由4个参数来定义:阳性对照(positive,p)和阴性对照(negative,n)的平均值(μ)和标准差(σ)。
计算公式如下:
z’factor=1-(3*(σp+σn)/|(μp-μn)|)
阴性对照组为加入溶剂(dmso)的未处理组;阳性对照用加入2.5μm的bez235或1μm的staurosporine。
一般细胞水平的功能性检测z’factor要求>0.3;本试验质控通过值z’factor设定为>0.5。
3.2活性数据
本发明10个化合物共用12个培养板。
培养板1~4用于细胞22rv1,其中培养板1用于式(5)所示的化合物、式(1)所示的化合物与式(3)所示化合物的活性检测,z'(bez)=0.76,z'(staurosporine)=0.80;培养板2用于式(2)所示化合物、式(4)所示化合物与式(10)所示化合物的活性检测,z'(bez)=0.75,z'(staurosporine)=0.79;培养板3用于式(7)所示化合物、式(8)所示化合物与式(6)所示化合物的活性检测,z'(bez)=0.75,z'(staurosporine)=0.79;培养板4用于式(9)所示化合物的活性检测,z'(bez)=0.82,z'(staurosporine)=0.85。
培养板5~8用于细胞22rv1-spp1,其中培养板5用于式(5)所示的化合物、式(1)所示的化合物与式(3)所示化合物的活性检测,z'(bez)=0.73,z'(staurosporine)=0.79;培养板6用于式(2)所示化合物、式(4)所示化合物与式(10)所示化合物的活性检测,z'(bez)=0.59,z'(staurosporine)=0.69;培养板7用于式(7)所示化合物、式(8)所示化合物与式(6)所示化合物的活性检测,z'(bez)=0.60,z'(staurosporine)=0.67;培养板8用于式(9)所示化合物的活性检测,z'(bez)=0.74,z'(staurosporine)=0.80。
其中,spp1(secretedphosphoprotein1),也称骨桥蛋白(osteopontin,opn)是一种分泌型sibling家族蛋白,在肿瘤细胞、巨噬细胞、破骨细胞,成纤维细胞,上皮细胞等多种细胞上表达。spp1主要是通过rgd(精氨酸-甘氨酸-天冬氨酸)及非rgd两个途径与细胞表面的整合素受体等相结合而实现其功能,与rgd基序相邻的是序列svvyglr,是隐藏的整合素结合位点:它在全长spp1中被隐藏,而肿瘤发生进展时,spp1被多种蛋白酶水解,暴露结合位点。spp1在介导肿瘤细胞粘附和迁移、骨组织的矿化与重建、免疫调节、血管新生、成纤维细胞激活与重编程等方面有着重要的作用。在前列腺癌和乳腺癌中,spp1被证明其高表达与前列腺癌的不良预后与生存期密切相关。
培养板9~12用于细胞lncap,其中培养板9用于式(5)所示的化合物、式(1)所示的化合物与式(3)所示化合物的活性检测,z'(bez)=0.65,z'(staurosporine)=0.70;培养板10用于式(2)所示化合物、式(4)所示化合物与式(10)所示化合物的活性检测,z'(bez)=0.74,z'(staurosporine)=0.78;培养板11用于式(7)所示化合物、式(8)所示化合物与式(6)所示化合物的活性检测,z'(bez)=0.62,z'(staurosporine)=0.70;培养板12用于式(9)所示化合物的活性检测,z'(bez)=0.76,z'(staurosporine)=0.79。
式(5)所示的化合物对各肿瘤细胞的活性曲线图如图21~图22所示,图21与图22中-19为式(5)所示的化合物的代号。由图21与图22可知,其对前列腺癌pc-3,du145,lncap,22rv1细胞均具有明显抗肿瘤活性,其中对黑色素瘤细胞wm115的抑制肿瘤效果最高。对卵巢癌细胞ovcar-4也要抑制作用。除此之外,对血管内皮细胞huvec-t11及微血管内皮细胞hmec-1也具有抑制作用。
图23为利用化学发光测定式(5)所示的化合物与恩杂鲁胺针对细胞22rv1联合用药得到的拟合药效计量曲线图。由图23可知,式(5)所示的化合物与恩杂鲁胺联合用药,针对耐药22rv1细胞有协同作用。
图24为式(1)所示的化合物(代号19-1)、式(3)所示化合物(代号19-2)、式(2)所示的化合物(代号19-3)、式(4)所示的化合物(代号19-4)与式(10)所示的化合物(代号19-5)针对肿瘤细胞22rv1的活性曲线图。
图25为式(7)所示化合物(代号19-6)、式(8)所示化合物(代号19-7)、式(6)所示化合物(代号19-8)与式(9)所示化合物(代号19-9)针对肿瘤细胞22rv1的活性曲线图。
图26为式(1)所示的化合物(代号19-1)、式(3)所示化合物(代号19-2)、式(2)所示的化合物(代号19-3)、式(4)所示的化合物(代号19-4)与式(10)所示的化合物(代号19-5)针对肿瘤细胞lncap的活性曲线图。
图27为式(7)所示化合物(代号19-6)、式(8)所示化合物(代号19-7)、式(6)所示化合物(代号19-8)与式(9)所示化合物(代号19-9)针对肿瘤细胞lncap的活性曲线图。
表1为实施例1制备的吲唑酰肼类化合物针对不同肿瘤细胞的活性数据。
表1抗肿瘤活性数据
- 还没有人留言评论。精彩留言会获得点赞!