N-保护的杂环类化合物、其制备方法及其用于制备C-核苷衍生物的方法与流程
本发明涉及化学合成领域,具体涉及n-保护的杂环类化合物、其制备方法及其用于制备c-核苷衍生物的方法。
背景技术:
c-核苷由于其较好成药性已经获得了广泛的研究和应用,一些c-核苷结构显示出较好的抗病毒或抗肿瘤药理活性。目前已经有c-核苷类似物的合成方法,然而一些特殊c-核苷的合成仍比较困难。
gilead公司于2012年报道了碱基1a和1c经临时硅保护后,可与核糖内酯进行加成和还原,得到c-核苷类似物3a和3c【bioorganic&medicinalchemistryletters2012,22(12),4127-4132;journalofmedicinalchemistry2014,57(5),1812-1825】,但从1a至3a或3c的收率不足20%。
gilead公司又于2017年报道了该方法的改进:从碱基1b出发,可以更可靠地获得c-核苷类似物3a【journalofmedicinalchemistry2017,60(5),1648-1661.】,但1b至3a的收率不足40%。
biotascientificmanagement和boehringeringelheim公司于2014年报道了c-核苷类似物3d和3e的更为复杂的合成方法【acsmedicinalchemistryletters2014,5(6),679-684】。
陈越磊等于2018年报道了一种改进方法,使用n-叔丁氧羰基保护的杂环类化合物制备c-核苷类似物3-1【organicchemistryfrontiers2018,5,1992-1999】,收率较上述文献有所提高。
然而,以上c-核苷类似物的合成方法仍有原料昂贵、收率较低或路线较长等问题。
因此,本领域急需提供合成路线短、成本低、产率高的c-核苷衍生物的制备方法。
技术实现要素:
本发明的目的就是提供合成路线短、成本低、产率高的c-核苷衍生物的制备方法。
本发明的另一目的在于提供一类c-核苷衍生物的中间体及其应用。
本发明的另一目的在于提供一种合成路线短、成本低、产率高的瑞德西韦的制备方法。
本发明第一方面,提供了一种式6化合物的制备方法,包括步骤:
(i)将式1化合物的氨基进行氨基保护反应,得到式2化合物;
(ii)式2化合物与式a化合物反应,得到式3化合物;
然后进行步骤(iii)或(iii'),
其中,(iii)式3化合物的羟基发生取代反应,得到式4化合物;和
(iii')式3化合物的羟基发生取代反应,得到式4a化合物;
以及步骤(iv)或(iv'),
其中,(iv)式4化合物发生完全脱保护反应,得到式6化合物;
(iv')式4a化合物发生完全脱保护反应,得到式6化合物;
上述各式中,
r选自下组:叔丁基或苄基;且
r1和r2独立地选自下组:取代或未取代的c6-c10芳基、取代或未取代的苄基、取代或未取代的萘甲基、取代或未取代的c1-c6烷基、取代或未取代的c1-c6烷基硅基,或取代或未取代的-si(ph)2-(c1-c6烷基);
其中,所述“取代”是指基团上的一个或多个(2、3、4、5个)氢被选自下组的基团取代:卤素、c1-c6烷基、c2-c6烯基、c2-c6炔基、c1-c6烷氧基、或c6-c10芳基。
在另一优选例中,在步骤(i)将式1化合物与式d1化合物进行反应,
式中,
r选自下组:叔丁基或苄基。
在另一优选例中,r1和r2独立地选自下组:2-萘甲基、1-萘甲基、叔丁基二甲基硅基、叔丁基二苯基硅基(tbdps)、三异丙基硅基、三乙基硅基、苄基、对甲氧基苄基、被卤素或c1-c6直链或支链烷基取代的苄基。
在另一优选例中,所述方法具有一个或多个下述特征:
(i)在惰性溶剂中,式1化合物的氨基进行氨基保护反应,得到式2化合物;
(ii)在惰性溶剂中,在有机锂和/或有机镁试剂存在下,式2化合物与式a化合物反应,得到式3化合物;
(iii)在惰性溶剂中,在酸和氰基化试剂存在下,式3化合物的羟基发生取代反应,得到式4化合物;或(iii')在惰性溶剂中,在酸和氰基化试剂存在下,式3化合物的羟基发生取代反应和氨基脱保护反应,得到式4a化合物;和/或
(iv)在惰性溶剂中,对所述式4化合物进行脱保护反应,得到式6化合物;或(iv')在惰性溶剂中,对所述式4a化合物进行脱保护反应,得到式6化合物。
在另一优选例中,所述式a化合物为
在另一优选例中,在步骤(iii')或(iii')中,所述酸独立地选自下组:tmsotf、三氟甲磺酸、三氟乙酸、三氟化硼-乙醚配合物、甲磺酸。
在另一优选例中,所述氰基化试剂选自下组:tmscn、tbscn、zncn,或其组合。
在另一优选例中,所述有机锂试剂为烷基锂。
在另一优选例中,所述有机锂试剂选自下组:甲基锂、仲丁基锂、叔丁基锂、lda,或其组合。
在另一优选例中,所述有机镁试剂选自下组:i-prmgcl·licl(turbogrignard试剂)、s-bumgcl·licl(另一种turbogrignard试剂)、tmpmgcl·licl(knochelhauserbase),或其组合。
本发明第二方面,还提供了一种式6化合物的制备方法,包括步骤:
(i)将式1化合物的氨基进行氨基保护反应,得到式2化合物;
(ii)式2化合物与式a化合物反应,得到式3化合物;
(iii)式3化合物的羟基发生取代反应,得到式4化合物;
(iv)式4化合物发生选择性脱保护反应,得到式5化合物;和
(v)式5化合物发生脱保护反应,得到式6化合物;和
上述各式中,
r选自下组:叔丁基或苄基;且
r1和r2独立地选自下组:取代或未取代的c6-c10芳基、取代或未取代的苄基、取代或未取代的萘甲基、取代或未取代的c1-c6烷基、取代或未取代的c1-c6烷基硅基,或取代或未取代的-si(ph)2-(c1-c6烷基);
其中,所述“取代”是指基团上的一个或多个(2、3、4、5个)氢被选自下组的基团取代:卤素、c1-c6烷基、c2-c6烯基、c2-c6炔基、c1-c6烷氧基、或c6-c10芳基。
在另一优选例中,所述方法具有一个或多个下述特征:
(i)在惰性溶剂中,式1化合物的氨基进行氨基保护反应,得到式2化合物;
(ii)在惰性溶剂中,在有机锂和/或有机镁存在下,式2化合物与式a化合物反应,得到式3化合物;
(iii)在惰性溶剂中,在酸和氰基化试剂存在下,式3化合物的羟基发生取代反应,得到式4化合物;
(iv)在惰性溶剂中,在路易斯酸(优选为三氯化硼)存在下,所述式4化合物发生选择性脱保护反应,得到式5化合物;和/或
(v)在惰性溶剂中,在路易斯酸或质子酸(优选为盐酸、三氟乙酸)存在下,所述式5化合物发生脱保护反应,得到式6化合物。
在另一优选例中,所述酸选自下组:tmsotf、三氟甲磺酸、三氟乙酸、三氟化硼-乙醚配合物、甲磺酸。
在另一优选例中,所述氰基化试剂选自下组:tmscn、tbscn、zncn,或其组合。
本发明第三方面,提供了一种瑞德西韦的制备方法,所述方法包括如本发明第一方面和本发明第二方面任一所述的制备化合物6的步骤。
在另一优选例中,所述方法还包括以下步骤:
本发明第四方面,还提供了一种瑞德西韦的制备方法,所述方法包括步骤:
(c)式9a化合物发生脱保护反应,得到式10化合物;
式中,r选自下组:叔丁基或苄基。
在另一优选例中,步骤(c)中,式9a化合物的羟基脱保护反应和氨基脱保护反应可以按任意顺序分步进行或同时进行。
在另一优选例中,在步骤(c)中,在惰性溶剂或无溶剂条件下,在脱保护剂存在下,式9a化合物发生脱保护反应,得到式10化合物。
在另一优选例中,所述脱保护剂选自下组:硫酸、盐酸、甲酸、醋酸、对甲苯磺酸、樟脑磺酸、甲磺酸、三氟乙酸、三氟甲磺酸。
在另一优选例中,在步骤(c)中,所述脱保护的温度为-20-50℃,较佳地,-10-30℃。
在另一优选例中,在步骤(c)中,所述惰性溶剂为水。
在另一优选例中,在步骤(c)之前,还包括步骤:
(b)式7a化合物与式8化合物发生反应,得到式9a化合物;
式中,lg选自下组:卤素、取代或未取代的c6-c10芳基氧基;
其中,所述“取代”是指基团中的一个或多个氢原子(1、2、3、4或5个)被选自下组的基团取代:卤素、硝基;且
r如上定义。
在另一优选例中,在步骤(b)中,在惰性溶剂中,在镁盐、锂盐和/或铝盐以及碱的存在下,式7a化合物与式8化合物发生反应,得到式9a化合物;或
在惰性溶剂中,在金属有机试剂存在下,式7a化合物与式8化合物发生反应,得到式9a化合物。
在另一优选例中,所述镁盐选自下组:氯化镁、溴化镁、碘化镁,或其组合。
在另一优选例中,所述锂盐选自下组:氯化锂、溴化锂、碘化锂,或其组合。
在另一优选例中,所述铝盐为三氯化铝。
在另一优选例中,所述碱为有机碱,优选地,二异丙基乙基胺、三乙胺、三丁基胺、n-甲基吗啉、n-甲基哌啶、吡啶、2,6-二甲基吡啶,或其组合。
在另一优选例中,所述金属有机试剂选自下组:叔丁基氯化镁、异丙基氯化镁、异丁基氯化镁、乙基氯化镁、丁基锂、钠氢、叔丁醇钾、叔丁醇钠、异戊醇钠,或其组合。
在另一优选例中,在步骤(b)之前,还包括步骤:
(a)式5化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,生成式7a化合物;
式中,r如上定义。
在另一优选例中,在步骤(a)中,在丙酮和/或2,2-二甲氧基丙烷中,在质子酸存在下,式5化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,生成式7a化合物。
在另一优选例中,所述质子酸选自下组:硫酸、对甲苯磺酸、樟脑磺酸、甲磺酸、三氟甲磺酸、三氟乙酸,或其组合。
在另一优选例中,在步骤(a)中,所述羟基保护反应的反应温度为-10-100℃,较佳地,20-50℃。
本发明第五方面,还提供了一种瑞德西韦的制备方法,其特征在于,所述方法包括步骤:
(z)式yy化合物发生脱保护反应,生成式10化合物;
式中,r选自下组:叔丁基或苄基。
在另一优选例中,在步骤(z)中,在惰性溶剂或无溶剂条件下,在脱保护剂存在下,式yy化合物发生脱保护反应,生成式10化合物。
在另一优选例中,在步骤(z)中,所述惰性溶剂为醇,较佳地,c1-c6烷基醇,更佳地,甲醇、乙醇、异丙醇,或其组合。
在另一优选例中,在步骤(z)中,所述惰性溶剂为醚类,较佳地,四氢呋喃、2-甲基四氢呋喃、甲基叔丁基醚、异丙醚、乙醚、乙二醇二甲醚,或其组合。
在另一优选例中,在步骤(z)中,所述惰性溶剂为卤代烃,较佳地,二氯甲烷、三氯甲烷、1,2-二氯乙烷,或其组合。
在另一优选例中,在步骤(z)中,所述惰性溶剂为芳香烃化合物,较佳地,甲苯、二甲苯、苯、氯苯,或其组合。
在另一优选例中,所述脱保护剂为质子酸,较佳地,所述质子酸选自下组:硫酸、盐酸、甲酸、醋酸、对甲苯磺酸、樟脑磺酸、甲磺酸、三氟乙酸、三氟甲磺酸,或其组合。
在另一优选例中,在步骤(z)中,所述惰性溶剂为水。
在另一优选例中,在步骤(z)之前,还包括步骤:
(y)式5化合物与式8化合物发生反应,生成式yy化合物;
式中,lg选自下组:卤素、取代或未取代的c6-c10芳基氧基;
其中,所述“取代”是指基团中的一个或多个氢原子(1、2、3、4或5个)被选自下组的基团取代:卤素、硝基;
r如上定义。
在另一优选例中,在步骤(y)中,在惰性溶剂中,在镁盐、锂盐和/或铝盐以及碱的存在下,式5化合物与式8化合物发生反应,得到式yy化合物;或
在惰性溶剂中,在金属有机试剂存在下,式5化合物与式8化合物发生反应,得到式yy化合物。
本发明第六方面,提供了一种式m化合物,
式中,
g选自下组:h或
其中,r选自下组:叔丁基或苄基;
r1和r2独立地选自下组:h、取代或未取代的c6-c10芳基、取代或未取代的苄基、取代或未取代的萘甲基、取代或未取代的c1-c6烷基、取代或未取代的c1-c6烷基硅基,取代或未取代的-si(ph)2-(c1-c6烷基),或两个r1一起形成-c(r4)2-;
r4独立地选自下组:取代或未取代的c1-c6烷基、取代或未取代的c2-c6烯基、取代或未取代的c2-c6炔基,或两个r4与其相连的c原子一起形成取代或未取代的4-7元碳环;
其中,所述“取代”是指基团上的一个或多个(2、3、4、5个)氢被选自下组的基团取代:卤素、c1-c6烷基、c2-c6烯基、c2-c6炔基、c1-c6烷氧基、或c6-c10芳基。
在另一优选例中,所述碳环为饱和碳环。
在另一优选例中,两个r4相同或不相同。
在另一优选例中,r4为甲基。
在另一优选例中,所述式m化合物选自下组:
各式中,r独立地选自下组:叔丁基或苄基。
本发明第七方面,提供了一种式m化合物的用途,用于制备c-核苷衍生物,或用作制备c-核苷衍生物的中间体。
在另一优选例中,所述c-核苷衍生物选自下组:式3化合物、式6化合物、式7化合物、式9化合物或瑞德西韦。
本发明第八方面,提供了瑞德西韦的制备方法,包括步骤:将式m化合物用作制备工艺的中间体或原料。
本发明第九方面,提供了一种式4化合物的制备方法,包括步骤:
将式3化合物的羟基发生取代反应,得到式4化合物;
式中,
r选自下组:叔丁基或苄基;且
r1和r2独立地选自下组:取代或未取代的c6-c10芳基、取代或未取代的苄基、取代或未取代的萘甲基、取代或未取代的c1-c6烷基、取代或未取代的c1-c6烷基硅基,或取代或未取代的-si(ph)2-(c1-c6烷基);
其中,所述“取代”是指基团上的一个或多个(2、3、4、5个)氢被选自下组的基团取代:卤素、c1-c6烷基、c2-c6烯基、c2-c6炔基、c1-c6烷氧基、或c6-c10芳基。
本发明第十方面,提供了一种式5化合物的制备方法,包括步骤:
将式4化合物发生选择性脱保护反应,得到式5化合物;和
式中,
r选自下组:叔丁基或苄基;且
r1和r2独立地选自下组:取代或未取代的c6-c10芳基、取代或未取代的苄基、取代或未取代的萘甲基、取代或未取代的c1-c6烷基、取代或未取代的c1-c6烷基硅基,或取代或未取代的-si(ph)2-(c1-c6烷基);
其中,所述“取代”是指基团上的一个或多个(2、3、4、5个)氢被选自下组的基团取代:卤素、c1-c6烷基、c2-c6烯基、c2-c6炔基、c1-c6烷氧基、或c6-c10芳基。
本发明第十一方面,提供了一种式7a化合物的制备方法,包括步骤:
式5化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,生成式7a化合物;
其中,r如上定义。
在另一优选例中,在步骤(a)中,在丙酮和/或2,2-二甲氧基丙烷中,在质子酸存在下,式5化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,生成式7a化合物。
在另一优选例中,所述质子酸选自下组:硫酸、盐酸、甲酸、醋酸、对甲苯磺酸、樟脑磺酸、甲磺酸、三氟乙酸、三氟甲磺酸,或其组合。
本发明第十二方面,提供了一种式9a化合物:
其中,r选自下组:叔丁基或苄基。
本发明第十三方面,提供了一种式9a化合物的制备方法,包括步骤:
(b)式7a化合物与式8化合物发生反应,得到式9a化合物;
式中,lg选自下组:卤素、取代或未取代的c6-c10芳基氧基;
其中,所述“取代”是指基团中的一个或多个氢原子(1、2、3、4或5个)被选自下组的基团取代:卤素、硝基;且
r如上定义。
在另一优选例中,lg选自下组:对硝基苯氧基、五氟苯氧基或氯。
在另一优选例中,在步骤(b)中,在惰性溶剂中,在镁盐、锂盐和/或铝盐以及碱的存在下,式7a化合物与式8化合物发生反应,得到式9a化合物;或
在惰性溶剂中,在金属有机试剂存在下,式7a化合物与式8化合物发生反应,得到式9a化合物。
本发明第十四方面,提供了一种式yy化合物:
其中,r选自下组:叔丁基或苄基。
本发明第十五方面,提供了一种式yy化合物的制备方法,包括步骤:
式5化合物与式8化合物发生反应,生成式yy化合物;
式中,lg选自下组:卤素、取代或未取代的c6-c10芳基氧基;
其中,所述“取代”是指基团中的一个或多个氢原子(1、2、3、4或5个)被选自下组的基团取代:卤素、硝基;
r如上定义。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
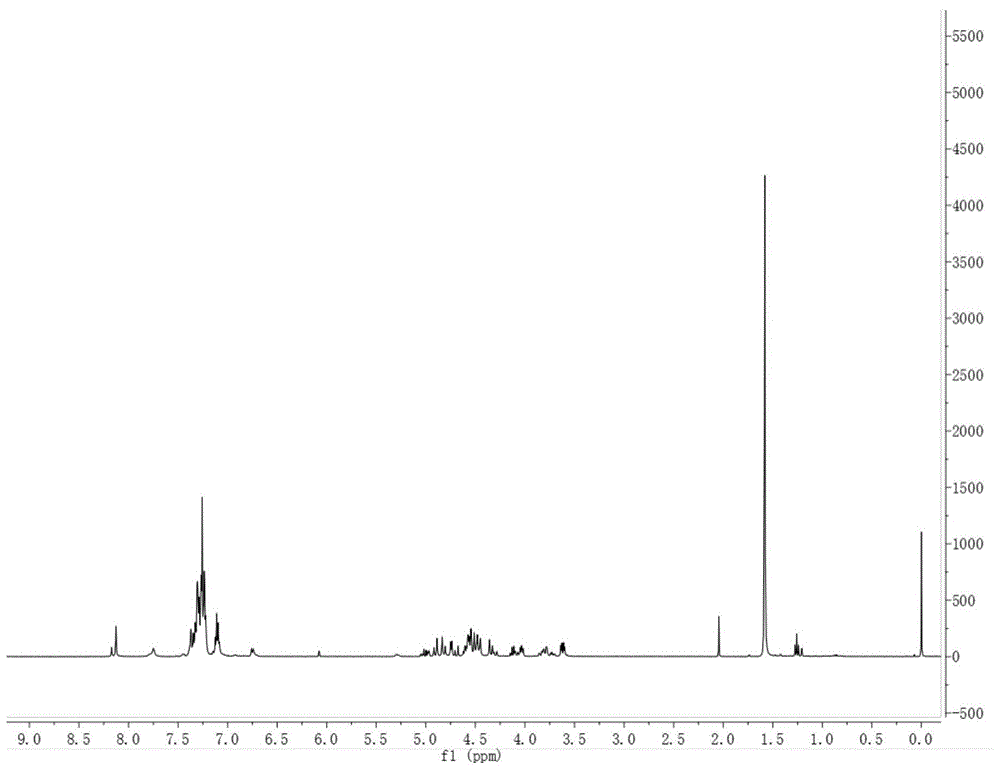
图1为实施例5制备的化合物4-1的1h-nmr谱图。
具体实施方式
本发明人经过广泛而深入的研究,通过大量筛选和测试,首次提供了一种使用n-苄氧羰基或n-叔丁氧羰基保护的杂环类化合物来制备c-核苷衍生物的方法。与现有技术相比,本发明无需进行氨基临时保护、或杂环卤代反应,即可直接使用正丁基锂脱除杂环类化合物的质子,与核糖内酯进行加成。令人惊讶地,本发明的方法不仅缩短了c-核苷衍生物的合成路线,而且在没有氨基临时保护或卤原子作为取代基的情况下,杂环类化合物与核糖内酯的反应获得了简便高效操作的特征、以及显著提高的收率;同时,氨基保护基(如boc)存在于合成路线后半部分,降低了化合物(如式5-1化合物)的极性,其在有机溶剂中溶解度更好,反应效率更高,且中间体纯化更为容易,产物纯度更高。在此基础上完成了本发明。
术语
除非另有定义,否则本文中所用的全部技术术语和科学术语均具有如本发明所属领域普通技术人员通常理解的相同含义。
如本文所用,在提到具体列举的数值中使用时,术语“约”意指该值可以从列举的值变动不多于1%。例如,如本文所用,表述“约100”包括99和101和之间的全部值(例如,99.1、99.2、99.3、99.4等)。
如本文所用,术语“含有”或“包括(包含)”可以是开放式、半封闭式和封闭式的。换言之,所述术语也包括“基本上由…构成”、或“由…构成”。
除非另有表述,术语“烷基”本身或作为另一取代基的一部分是指具有指定碳原子数的直链或支链烃基(即,c1-6表示1-6个碳),烷基可包括1、2、3、4、5或6碳原子的烷基。烷基的例子包括甲基、乙基、正丙基、异丙基、正丁基、叔丁基、异丁基、仲丁基、正戊基、正己基等。
除非另有表述,术语“烯基”指具有一个或多个双键的不饱和烷基,烯基可包括2、3、4、5或6碳原子的烯基。类似地,术语“炔基”指具有一个或多个三键的不饱和烷基,炔基可包括2、3、4、5或6碳原子的炔基。此类不饱和烷基的例子包括乙烯基、2-丙烯基、巴豆基、2-异戊烯基、2-(丁二烯基)、2,4-戊二烯基、3-(1,4-戊二烯基)、乙炔基、1-和3-丙炔基、3-丁炔基和更高级的同系物和异构体。
除非另有表述,术语“卤代”或“卤素”本身或作为另一取代基的一部分是指氟、氯、溴、或碘原子。此外,诸如“卤代烷基”等术语表示包括单卤代烷基或多卤代烷基。例如,术语“c1-4卤代烷基”表示包括三氟甲基、2,2,2-三氟乙基、4-氯丁基、3-溴丙基等。
除非另有表述,术语“芳基”表示多不饱和的(通常芳香性)的烃基,其可以是单环或稠合在一起或共价连接的多环(最多三环)。例如,芳基的例子包括苯基、萘基。
如本文所用,“惰性溶剂”是指甲醇、乙醇、异丙醇、氯仿、苯、二甲亚砜、n-甲基吡咯烷酮、n、n-二甲基甲酰胺、丙酮、乙腈、醋酸、甲酸、正己烷、正庚烷、甲苯、四氢呋喃、乙酸乙酯、1,4-二氧六环、甲基叔丁基醚、水或上述溶剂的混合物。
如本文所用,“式10化合物”与“瑞德西韦”可互换使用,指具有式10所示结构的化合物或其药学上可接受的盐。
如本文所用,
如本文所用,“室温”指4-40℃,较佳地,25±5℃。
制备方法
下面更具体地描述本发明的式2、4、5以及6的化合物的制法以及进一步制备瑞德西韦的方法,但这些具体方法不对本发明构成任何限制。基于本发明的教导,式6化合物以及瑞德西韦还可以任选将在本说明书中描述的方法和本领域已知的各种合成方法组合起来而方便地制得,这样的组合可由本发明所属领域的技术人员容易地进行。
本发明的方法中的反应可以在任何合适的温度下进行。例如,反应温度可以是约-78℃至约100℃,或者为约-50℃至约100℃,或者为约-25℃至约50℃,或约-10℃至约25℃,或约0℃至约20℃。在一些实施方案中,反应温度可以是约0℃至约20℃。
通常,在制备流程中,各反应通常在惰性溶剂中,在室温至回流温度(如0℃~100℃,优选0℃~80℃)下进行。反应时间通常为0.1小时-60小时,较佳地为0.5-48小时,如1、2、6、12、18、24或36h。
下面的通用制备路线可以用于合成本发明式2、3、4、5、6的化合物以及用于制备瑞德西韦。
本发明提供了一种式m化合物,
式中,
g选自下组:h或
其中,r选自下组:叔丁基或苄基;
r1和r2独立地选自下组:h、取代或未取代的c6-c10芳基、取代或未取代的苄基、取代或未取代的萘甲基、取代或未取代的c1-c6烷基、取代或未取代的c1-c6烷基硅基,取代或未取代的-si(ph)2-(c1-c6烷基),或两个r1一起形成-c(r4)2-;
r4独立地选自下组:取代或未取代的c1-c6烷基、取代或未取代的c2-c6烯基、取代或未取代的c2-c6炔基,或两个r4与其相连的c原子一起形成取代或未取代的4-7元碳环;
其中,所述“取代”是指基团上的一个或多个(2、3、4、5个)氢被选自下组的基团取代:卤素、c1-c6烷基、c2-c6烯基、c2-c6炔基、c1-c6烷氧基、或c6-c10芳基。
在另一优选例中,所述式m化合物选自下组:
各式中,r独立地选自下组:叔丁基或苄基。
式2化合物的制备
本发明中,式2化合物可通过式1化合物制备:
优选地,在该步骤中,将式1化合物与式d1化合物进行反应,从而制得式2化合物
式中,
各r独立地选自下组:叔丁基、苄基。
式3化合物的制备
优选地,在该步骤中,将式2化合物与式a化合物进行反应,从而制得式3化合物。
优选地,在该步骤中,所述反应在有机锂和/或有机镁试剂存在下进行。
在另一优选例中,所述有机锂试剂为烷基锂。
在另一优选例中,所述有机锂试剂选自下组:甲基锂、仲丁基锂、叔丁基锂、lda,或其组合。
在另一优选例中,所述有机镁试剂选自下组:i-prmgcl·licl(turbogrignard试剂)、s-bumgcl·licl(另一种turbogrignard试剂)、tmpmgcl·licl(knochelhauserbase),或其组合。
优选地,所述式a化合物为
式4化合物的制备
式3化合物的羟基发生取代反应,得到式4化合物
优选地,所述步骤中,式3化合物在酸和氰基化试剂存在下,羟基被氰基取代。
在另一优选例中,所述酸选自下组:tmsotf、三氟甲磺酸、三氟乙酸、三氟化硼-乙醚配合物、甲磺酸或其组合。
在另一优选例中,所述氰基化试剂选自下组:tmscn、tbscn、zncn,或其组合。
式5化合物的制备
式4化合物发生选择性脱保护反应,得到式5化合物
优选地,所述选择性脱保护在路易斯酸存在下进行,更佳地,三氯化硼。
式6化合物的制备
或者,当两个r1均为h,且r2为h时,可从式5化合物制备式6化合物。
优选地,式6化合物可通过下述方法制备:
优选地,上述各式中,
r选自下组:叔丁基或苄基;且
r1和r2独立地选自下组:取代或未取代的c6-c10芳基、取代或未取代的苄基、取代或未取代的萘甲基、取代或未取代的c1-c6烷基、取代或未取代的c1-c6烷基硅基,或取代或未取代的-si(ph)2-(c1-c6烷基);
其中,所述“取代”是指基团上的一个或多个(2、3、4、5个)氢被选自下组的基团取代:卤素、c1-c6烷基、c2-c6烯基、c2-c6炔基、c1-c6烷氧基、或c6-c10芳基。
本发明还提供另一种从式3化合物制备式6化合物的方法,包括步骤:
(iii')式3化合物的羟基发生取代反应,得到式4a化合物;和
(iv')式4a化合物发生完全脱保护反应,得到式6化合物;
式中,r、r1、r2如上定义。
在另一优选例中,在式(iii')中,在惰性溶剂中,在酸和氰基化试剂存在下,式3化合物的羟基发生取代反应和氨基脱保护反应,得到式4a化合物。
在另一优选例中,在式(iv')中,在惰性溶剂中,对所述式4a化合物进行脱保护反应,得到式6化合物。
式7a化合物的制备方法
本发明提供了一种式7a化合物的制备方法,包括步骤:
(a)式5化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,生成式7a化合物;
其中,r如上定义。
在另一优选例中,在步骤(a)中,在丙酮和/或2,2-二甲氧基丙烷中,在质子酸存在下,式5化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,生成式7a化合物。
在另一优选例中,所述质子酸选自下组:硫酸、对甲苯磺酸、樟脑磺酸、甲磺酸、三氟甲磺酸、三氟乙酸,或其组合。
在另一优选例中,在步骤(a)中,所述羟基保护反应的反应温度为-40-100℃,较佳地,-20~-10℃。
式7b化合物的制备方法
本发明提供了一种式7b化合物的制备方法,包括步骤:
(a)式5-1化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,同时发生脱保护反应,生成式7b化合物;
在另一优选例中,在步骤(a)中,在丙酮和/或2,2-二甲氧基丙烷中,在质子酸存在下,式5-1化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,生成式7b化合物。
在另一优选例中,所述质子酸选自下组:硫酸、对甲苯磺酸、樟脑磺酸、甲磺酸、三氟甲磺酸、三氟乙酸,或其组合。
在另一优选例中,在步骤(a)中,所述羟基保护反应的反应温度为-40-100℃,较佳地,20~40℃。
式9a化合物
式中,r选自下组:叔丁基或苄基。
式9a化合物的制备方法
本发明提供了一种式9a化合物的制备方法,包括步骤:
(b)式7a化合物与式8化合物发生反应,得到式9a化合物;
式中,lg选自下组:卤素、取代或未取代的c6-c10芳基氧基;
其中,所述“取代”是指基团中的一个或多个氢原子(1、2、3、4或5个)被选自下组的基团取代:卤素、硝基;且
r如上定义。
在另一优选例中,lg选自下组:对硝基苯氧基、五氟苯氧基或氯。
在另一优选例中,在步骤(b)中,在惰性溶剂中,在镁盐、锂盐和/或铝盐以及碱的存在下,式7a化合物与式8化合物发生反应,得到式9a化合物;或
在惰性溶剂中,在金属有机试剂存在下,式7a化合物与式8化合物发生反应,得到式9a化合物。
在另一优选例中,所述镁盐选自下组:氯化镁、溴化镁、碘化镁,或其组合。
在另一优选例中,所述锂盐选自下组:氯化锂、溴化锂、碘化锂,或其组合。
在另一优选例中,所述铝盐为三氯化铝。
在另一优选例中,所述碱为有机碱,优选地,二异丙基乙基胺、三乙胺、三丁基胺、n-甲基吗啉、n-甲基哌啶、吡啶、2,6-二甲基吡啶,或其组合。
在另一优选例中,所述金属有机试剂选自下组:叔丁基氯化镁、异丙基氯化镁、异丁基氯化镁、乙基氯化镁、丁基锂、钠氢、叔丁醇钾、叔丁醇钠、异戊醇钠,或其组合。
式yy化合物
其中,r选自下组:叔丁基或苄基。
式yy化合物的制备方法
式yy化合物的制备方法,包括步骤:
式5化合物与式8化合物发生反应,生成式yy化合物;
式中,lg选自下组:卤素、取代或未取代的c6-c10芳基氧基;
其中,所述“取代”是指基团中的一个或多个氢原子(1、2、3、4或5个)被选自下组的基团取代:卤素、硝基;
r如上定义。
瑞德西韦的制备
本发明的式m化合物可用于制备瑞德西韦或作为制备瑞德西韦的中间体。
本发明提供了一种瑞德西韦的制备的制备方法,包括步骤:将式m化合物用作瑞德西韦制备工艺的中间体或原料。
典型地,式6化合物为抗病毒药物瑞德西韦(式10化合物)的关键中间体,所以通过本发明的方法或本发明的新颖中间体,可进一步制备瑞德西韦。
优选地,一种制备瑞德西韦的方法包括上述的制备式6化合物的步骤。
优选地,一种制备瑞德西韦的方法还可包括额外的从式6化合物制备瑞德西韦的步骤。
典型地,一种从式6化合物制备瑞德西韦的方法包括步骤:
其中,包括:
对式6化合物用保护基进行基团保护,从而获得式7化合物;
将式7化合物与式8化合物进行反应,从而获得式9化合物;
对式9化合物进行脱保护基反应,从而获得式10化合物(瑞德西韦)。
本发明还提供了一种瑞德西韦的制备方法,包括步骤:
(c)式9a化合物发生脱保护反应,得到式10化合物;
式中,r选自下组:叔丁基或苄基。
在另一优选例中,步骤(c)中,式9a化合物的羟基脱保护反应和氨基脱保护反应可以按任意顺序分步进行或同时进行。
在另一优选例中,在步骤(c)中,在惰性溶剂或无溶剂条件下,在脱保护剂存在下,式9a化合物发生脱保护反应,得到式10化合物。在另一优选例中,所述脱保护剂选自下组:硫酸、盐酸、甲酸、醋酸、对甲苯磺酸、樟脑磺酸、甲磺酸、三氟乙酸、三氟甲磺酸。
在另一优选例中,在步骤(c)中,所述脱保护的温度为-20-50℃,较佳地,-10-30℃。
在另一优选例中,在步骤(c)之前,还包括步骤:
(b)式7a化合物与式8化合物发生反应,得到式9a化合物;
式中,lg选自下组:卤素、取代或未取代的c6-c10芳基氧基;
其中,所述“取代”是指基团中的一个或多个氢原子(1、2、3、4或5个)被选自下组的基团取代:卤素、硝基;
r如上定义。
在另一优选例中,在步骤(b)中,在惰性溶剂中,在镁盐、锂盐和/或铝盐以及碱的存在下,式7a化合物与式8化合物发生反应,得到式9a化合物;或
在惰性溶剂中,在金属有机试剂存在下,式7a化合物与式8化合物发生反应,得到式9a化合物。
在另一优选例中,在步骤(b)之前,还包括步骤:
(a)式5化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,生成式7a化合物;
式中,r如上定义。
在另一优选例中,在步骤(a)中,在丙酮和/或2,2-二甲氧基丙烷中,在质子酸存在下,式5化合物与丙酮和/或2,2-二甲氧基丙烷发生羟基保护反应,生成式7a化合物。
优选地,本发明的制备瑞德西韦的方法包括如下步骤:
式中,r和lg如上定义。
本发明还提供了一种瑞德西韦的制备方法,包括步骤:
(z)式yy化合物发生脱保护反应,生成式10化合物;
式中,r选自下组:叔丁基或苄基。
在另一优选例中,在步骤(z)中,在惰性溶剂中,在脱保护剂存在下,式yy化合物发生脱保护反应,生成式10化合物。
在另一优选例中,在步骤(z)中,所述惰性溶剂为醇,较佳地,c1-c6烷基醇,更佳地,甲醇、乙醇、异丙醇,或其组合。
在另一优选例中,所述脱保护剂为质子酸,较佳地,所述质子酸选自下组:硫酸、对甲苯磺酸、樟脑磺酸、甲磺酸、三氟甲磺酸、三氟乙酸,或其组合。
在另一优选例中,在步骤(z)之前,还包括步骤:
(y)式5化合物与式8化合物发生反应,生成式yy化合物;
式中,lg选自下组:卤素、取代或未取代的c6-c10芳基氧基;
其中,所述“取代”是指基团中的一个或多个氢原子(1、2、3、4或5个)被选自下组的基团取代:卤素、硝基;
r如上定义。
在另一优选例中,在步骤(y)中,在惰性溶剂中,在镁盐、锂盐和/或铝盐以及碱的存在下,式5化合物与式8化合物发生反应,得到式yy化合物;或
在惰性溶剂中,在金属有机试剂存在下,式5化合物与式8化合物发生反应,得到式yy化合物。
优选地,本发明的制备瑞德西韦的方法包括如下步骤:
式中,r和lg如上定义。
在另一优选例中,r为叔丁基。
令人惊讶地,当化合物5、7a、9a和yy中氨基被叔丁基氧羰基(boc)或苄氧羰基(cbz)保护后,化合物具有较低的极性,使得产品易于纯化,显著提高了产物的收率和纯度。例如,当化合物5的r基团为boc基团时,在有机溶剂中溶解度更好,因此和式8化合物反应收率显著提高。
还可从式7b化合物经由9b制备瑞德西韦。
本发明的主要优点包括:
(a)本发明从易于制备的、成本较低的化合物2出发合成化合物6,因此成本低,并避免使用昂贵的卤化物原料;
(b)本发明避免使用卤代化合物2对核糖内酯a加成时必须的临时氨基保护,因此反应的可重复性有明显提高;
(c)本发明方法的收率高;
(d)本发明方法的立体选择性好;
(e)本发明易于工业化生产等优势;
(f)当化合物5的氨基具有boc或cbz保护基团时,在有机溶剂中溶解度更好,因此和式8化合物反应收率显著提高。
(g)本发明制备瑞德西韦的方法中,中间体易纯化,产物的收率和纯度显著提高。
下面结合具体实施,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
缩写:
tlc为薄层色谱;
etoac为乙酸乙酯;
thf为四氢呋喃;
tbme为叔丁基甲基醚;
boc2o为二碳酸二叔丁酯;
cbz2o为苄氧甲酸酐;
cbzcl为氯甲酸苄酯;
lc-ms在agilent1260infinityⅱ+infinitylablc/msd上测量。tlc在wfh-203b紫外灯下检查。高分辨质谱在waters,q-tofultimaglobalspectrometer上测量。核磁在布鲁克400m核磁仪上测量。
液相色谱在agilent1260infinityⅱ,thermou3000,或shimadzulc-16仪器上测量。液相方法为:
色谱柱:welchcn(250mm×4.6mm×5um)
流动相:a——0.1%磷酸水;b——0.1%磷酸乙腈。
流速:1.0ml/min。
柱温:40℃。
检测波长:210nm。
进样体积:1μl。
分析时间:40min
梯度程序:
或:
色谱柱:welchc18(250mm×4.6mm×5um)。
流动相:a——0.1%磷酸水;b——0.1%磷酸甲醇。
流速:1.0ml/min。
柱温:40℃。
检测波长:240nm。
进样体积:1μl。
分析时间:40min
梯度程序:
实施例1化合物2-1的制备
反应瓶中加入1.34g吡咯并[2,1-f][1,2,4]三嗪-4-胺(化合物1),20ml四氢呋喃和20ml饱和碳酸氢钠水溶液,再分批加入3.27gboc2o,在20~25℃下反应16小时,加入50ml水,用乙酸乙酯(20ml×2)萃取,合并有机相,用饱和食盐水洗涤(30ml×3),无水硫酸钠干燥,浓缩,硅胶柱分离(10:1,v/v,石油醚:乙酸乙酯),得2.13g白色固体产物2-1,产率:91%。hrms(esi+):c11h13n4nao2+,m.w.256.0935。
实施例2化合物2-2的制备
向反应瓶中加入化合物1(1.2mmol),dmf(10ml)和三乙胺(2.0mmol),滴加苄氧羰基氯(2.0mmol),在40~25℃下反应16小时,加入50ml水,用乙酸乙酯(20ml×2)萃取,合并有机相,用饱和食盐水洗涤(30ml×3),无水硫酸钠干燥,浓缩,柱层析分离得白色固体产物2-2(0.5mmol)。hrms(esi+):c14h11n4nao2+,m.w.290.0776。
实施例3化合物3-1的制备
将化合物2-1(1.0mmol)溶于thf(7ml)中,冷却至-50~-70℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。向此混合物加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)在thf(10ml)中的溶液,并将此混合物在-50℃搅拌5h,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物3-1(0.69mmol),hrms(esi+):c37h40n4nao7+,m.w.675.2799。
实施例3.1化合物3-1的制备
将化合物2-1(1.0mmol)溶于thf(7ml)中,冷却至-50~-70℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,并将上述锂化混合物加入此反应液中。将此混合物继续在-50℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物3-1(0.65mmol),hrms分析结果与实施例3所得化合物3-1样品一致。
实施例3.2化合物3-1的制备
将化合物2-1(1.0mmol)溶于thf(7ml)中,冷却至-50~-70℃,先滴加异丙胺(0.01mmol),再滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,并将上述锂化混合物加入此反应液中。将此混合物继续在-50℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物3-1(0.66mmol),hrms分析结果与实施例3所得化合物3-1样品一致。
实施例4化合物3-2的制备
将化合物2-2(1.0mmol)溶于thf(7ml)中,冷却至-50~-70℃,先滴加异丙胺(0.01mmol),再滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,并将上述锂化混合物加入此反应液中。将此混合物继续在-50℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物3-2(0.41mmol),hrms(esi+):c40h38n4nao7+,m.w.709.2639。
实施例5化合物4-1的制备
将化合物3-1(2.3mmol)在dcm(50ml)中的溶液冷却至-40℃,加入三氟乙酸(6mmol),再加入tmsotf(10mmol)和tmscn(10mmol)在dcm(20ml)中预冷至-40℃的混合溶液,随后将此混合物在-30℃搅拌30min,并加入预冷至-10℃的koh水溶液中终止,将此混合物分液后,有机相用饱和食盐水洗涤、浓缩。最后将所得有机相浓缩后柱层析得到产物4-1(2.0mmol),hrms(esi+):c38h39n5nao6+,m.w.684.2810。核磁谱图图1所示。
实施例6化合物4-1的制备
采用类似于实施例5中的方法,不同点在于,采用zn(cn)2替换tmscn。
将化合物3-1(2.3mmol)和zn(cn)2(1.5mmol)在dcm(50ml)中的悬浊液冷却至-40℃,加入三氟乙酸(6mmol),再加入tmsotf(10mmol),随后将此混合物在-30℃搅拌30min,并加入饱和醋酸钠水溶液中终止,将此混合物分液后,有机相用饱和食盐水洗涤、浓缩。最后将所得有机相浓缩后柱层析得到产物4-1(0.21mmol),hrms分析结果与实施例5所得化合物4-1样品一致。
实施例7化合物4-2的制备
将化合物3-2(2.5mmol)在dcm(50ml)中的溶液冷却至-40℃,加入三氟乙酸(6mmol),再加入tmsotf(10mmol)和tmscn(10mmol)在dcm(20ml)中预冷至-40℃的混合溶液,随后将此混合物在-30℃搅拌30min,并加入预冷至-10℃的koh水溶液中终止,将此混合物分液后,有机相用饱和食盐水洗涤、浓缩。最后将所得有机相浓缩后柱层析得到产物4-2(2.1mmol),hrms(esi+):c41h37n5nao6+,m.w.718.2645。
实施例8化合物4-2的制备
采用类似于实施例7中的方法,不同点在于,采用zn(cn)2替换tmscn。
实施例9化合物4a-1的制备
将化合物3-1(2.3mmol)在dcm(50ml)中的溶液冷却至-40℃,加入三氟甲磺酸(6mmol),再加入tmsotf(10mmol)和tmscn(10mmol)在dcm(20ml)中预冷至-40℃的混合溶液,随后将此混合物在-30℃搅拌30min,并加入预冷至-10℃的koh水溶液中终止,将此混合物分液后,有机相用饱和食盐水洗涤、浓缩。最后将所得有机相浓缩后柱层析得到产物4a-1(2.1mmol),hrms(esi+):c33h31n5nao4+,m.w.584.2272。
实施例10化合物5-1的制备
将化合物4-1(1mmol)溶于dcm(5ml)中冷却至-60℃,加入三氯化硼(1mdcm溶液,3.4mmol),继续在-60℃搅拌3h,随后加入甲醇(30ml),将此混合物在-10度以下浓缩至无溶剂流出,再加甲醇(30ml)浓缩至无溶剂流出,随后将此混合物加入碳酸钾水溶液(20%),逐步热至室温,析出固体,过滤,水洗,并干燥,得到产物5-1(0.85mmol),hrms(esi+):c17h21n5nao6+,m.w.414.1389。
实施例11化合物6的制备
将化合物5-1(1mmol)溶于2mlhcl-异丙醇溶液(1m),室温搅拌1h,过滤,得到产物6的盐酸盐(0.95mmol),hrms(esi+):c12h14n5o4+,m.w.292.1048。
实施例12化合物6的制备
将化合物4-1(1mmol)溶于dcm(5ml)中冷却至-20℃,加入三氯化硼(1mdcm溶液,4mmol),继续在-20℃搅拌3h,随后加入甲醇(30ml),将此混合物在-10度以下浓缩至无溶剂流出,再加甲醇(30ml)浓缩至无溶剂流出,随后将此混合物加入碳酸钾水溶液(20%),逐步热至室温,析出固体,过滤,水洗,并干燥,得到产物6(0.91mmol),hrms分析结果与实施例11所得化合物6样品一致。
实施例13化合物6的制备
将化合物4-2(1mmol)溶于dcm(5ml)中冷却至-20℃,加入三氯化硼(1mdcm溶液,4mmol),继续在-20℃搅拌3h,再在-10℃搅拌3h,随后加入甲醇(30ml),将此混合物在-10度以下浓缩至无溶剂流出,再加甲醇(30ml)浓缩至无溶剂流出,随后将此混合物加入碳酸钾水溶液(20%),逐步热至室温,析出固体,过滤,水洗,并干燥,得到产物6(0.65mmol),hrms分析结果与实施例11所得化合物6样品一致。
实施例14化合物5-2的制备
将化合物4-2(1mmol)溶于dcm(5ml)中冷却至-60℃,加入三氯化硼(1mdcm溶液,3.4mmol),继续在-60℃搅拌3h,随后加入甲醇(30ml),将此混合物在-10度以下浓缩至无溶剂流出,再加甲醇(30ml)浓缩至无溶剂流出,随后将此混合物加入碳酸钾水溶液(20%),逐步热至室温,析出固体,过滤,水洗,并干燥,得到产物5-2(0.85mmol),hrms(esi+):c20h20n5o6+,m.w.426.1415。
实施例15化合物7a-1的制备
将化合物5-1(2mmol)溶于丙酮(10ml)和2,2-二甲氧基丙烷(5ml)中,用无水对甲苯磺酸调ph至1,在-10~-20℃之间搅拌2h,加碳酸氢钠调节反应体系ph至7以上,将反应混合物浓缩干,残留物柱层析得7a-1(1.84mmol)。hrms(esi+):c20h25n5nao6+,m.w.454.1704。
实施例15.1化合物7b的制备
将化合物5-1(2mmol)溶于丙酮(10ml)和2,2-二甲氧基丙烷(5ml)中,用无水对甲苯磺酸调ph至1,在20~30℃之间搅拌2h,加碳酸氢钠调节反应体系ph至7以上,将反应混合物浓缩干,残留物柱层析得7b(1.9mmol)。hrms(esi+):c15h17n5nao4+,m.w.354.1178。
实施例16化合物7a-2的制备
将化合物5-1(2mmol)溶于丙酮(10ml)和2,2-二甲氧基丙烷(5ml)中,用无水对甲苯磺酸调ph至1,室温搅拌2h,加碳酸氢钠调节反应体系ph至7以上,将反应混合物浓缩干,残留物柱层析得7a-2(1.9mmol)。hrms(esi+):c23h23n5nao6+,m.w.488.1549。
实施例17化合物9a-1的制备
将化合物7a-1(2mmol),化合物8(2.1mmol),氯化镁(2.3mmol)悬浮于无水乙腈(30ml)中,滴加dipea(4mmol),将混合物在0~10℃搅拌3h,随后加入2-甲基四氢呋喃(20ml)和水(20ml),分液,将有机相依次用柠檬酸水溶液,碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到9a-1(浅黄色油状物,1.8mmol)。hrms(esi+):c35h47n6nao10p+,m.w.765.2990。
实施例17.1化合物9b的制备
将化合物7b(2mmol),化合物8(2.1mmol),氯化镁(2.3mmol)悬浮于无水乙腈(30ml)中,滴加dipea(4mmol),将混合物在0~10℃搅拌3h,随后加入2-甲基四氢呋喃(20ml)和水(20ml),分液,将有机相依次用柠檬酸水溶液,碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到9b(1.6mmol)。hrms(esi+):c30h39n6nao8p+,m.w.665.2462。
实施例18化合物9a-2的制备
将化合物7a-2(2mmol),化合物8(2.1mmol),氯化镁(2.3mmol)悬浮于无水乙腈(40ml)中,滴加dipea(4mmol),将混合物在0~10℃搅拌3h,随后加入2-甲基四氢呋喃(30ml)和水(30ml),分液,将有机相依次用柠檬酸水溶液,碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到9a-2(无色油状物,1.8mmol)。hrms(esi+):c38h45n6nao10p+,m.w.799.2832。
实施例19化合物9a-2的制备
将化合物7a-2(2mmol),化合物8(2.1mmol),氯化镁(2.3mmol)悬浮于无水乙腈(40ml)中,滴加dipea(4mmol),将混合物在0~10℃搅拌3h,随后加入2-甲基四氢呋喃(10ml)和水(10ml),分液,将有机相依次用柠檬酸水溶液,碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到9a-2(无色油状物,1.6mmol)。分析数据与9a-2标准样品一致。hrms分析结果与实施例18所得化合物9a-2样品一致。
实施例20瑞德西韦的制备
将化合物9b(1mmol)溶于乙腈(20ml),0℃下加入浓盐酸(5ml),0~10℃搅拌2h,随后加入2-甲基四氢呋喃(30ml)和水(30ml),分液,将有机相依次用碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到瑞德西韦10(0.8mmol)。hrms(esi+):c27h35n6nao8p+,m.w.625.2153。
实施例21瑞德西韦的制备
将化合物9a-1(1mmol)溶于乙腈(20ml),0℃下加入浓盐酸(5ml),0~10℃搅拌2h,随后加入2-甲基四氢呋喃(30ml)和水(30ml),分液,将有机相依次用碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到瑞德西韦10(0.8mmol)。hrms(esi+):c27h35n6nao8p+,m.w.625.2153。
实施例22瑞德西韦的制备
将化合物9a-2(1mmol)溶于乙腈(20ml),0℃下加入浓盐酸(5ml),室温搅拌2h,随后加入2-甲基四氢呋喃(30ml)和水(30ml),分液,将有机相依次用碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到瑞德西韦10(0.2mmol)。hrms分析结果与实施例21所得化合物10样品一致。
实施例23化合物yy的制备
将化合物5-1(1mmol),化合物8(1.1mmol),氯化镁(1.2mmol)悬浮于无水乙腈(20ml)中,滴加dipea(2mmol),将混合物在0~10℃搅拌3h,随后加入2-甲基四氢呋喃(10ml)和水(10ml),分液,将有机相依次用柠檬酸水溶液,碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到yy(0.7mmol)。hrms(esi+):c32h43n6nao10p+,m.w.725.2677。
实施例24化合物yy的制备
将化合物5-1(1mmol),化合物9(1.1mmol),氯化镁(1.2mmol)悬浮于无水乙腈(20ml)中,滴加dipea(2mmol),将混合物在0~10℃搅拌3h,随后加入2-甲基四氢呋喃(10ml)和水(10ml),分液,将有机相依次用柠檬酸水溶液,碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到yy(0.8mmol)。hrms分析结果与实施例23所得化合物yy样品一致。
实施例25瑞德西韦的制备
将化合物yy(1mmol)溶于乙腈(20ml),0℃下加入浓盐酸(5ml),0~10℃搅拌2h,随后加入2-甲基四氢呋喃(30ml)和水(30ml),分液,将有机相依次用碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到10(0.9mmol)。hrms分析结果与实施例21所得化合物10样品一致。
实施例26化合物yy的制备
将化合物5-2(1mmol),化合物8(1.1mmol),氯化镁(1.2mmol)悬浮于无水乙腈(20ml)中,滴加dipea(2mmol),将混合物在0~10℃搅拌3h,随后加入2-甲基四氢呋喃(10ml)和水(10ml),分液,将有机相依次用柠檬酸水溶液,碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到yya(0.8mmol)。hrms(esi+):c35h41n6nao10p+,m.w.759.2516。
实施例27瑞德西韦的制备
将化合物yya(1mmol)溶于乙腈(20ml),0℃下加入浓盐酸(5ml),室温搅拌2h,随后加入2-甲基四氢呋喃(30ml)和水(30ml),分液,将有机相依次用碳酸钾水溶液和水洗涤,将有机相浓缩干,残余物柱层析得到10(0.3mmol)。hrms分析结果与实施例21所得化合物10样品一致。
结果表明,与现有技术相比,在本发明方法中,对杂环类化合物仅进行n的保护而无需进行卤代(溴代或碘代),无需使用氨基临时保护,然后直接使用有机锂或有机镁脱除质子,与核糖内酯进行加成,不仅缩短了路线,而且提高了收率。本发明人出乎意料的发现,式5化合物的氨基具有boc或cbz保护基时,式5化合物在有机溶剂中具有良好的溶解性,可以直接与膦酰胺(式8化合物)反应得到结构yy,从而避免额外的使用丙酮保护的步骤。且式5化合物的氨基具有boc或cbz保护基时,各中间体易于纯化,产物的产率和纯度显著提高。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!