热激活延迟红光荧光材料、有机电致发光器件与显示装置
本发明涉及有机光电材料技术领域,尤其涉及一种热激活延迟红光荧光材料、有机电致发光器件与显示装置。
背景技术:
有机电致发光器件(organiclightemittingdiodes),简称oled,是一类利用有机材料,在电场作用下,受电流或电压激发而产生光的器件,多采用三明治结构。oled以其饱和的色彩质量、低功耗、响应速度快、可大面积制备等不可比拟的优势,在新型显示和照明技术领域备受关注。目前,oled主要应用于电脑和手机的显示屏幕领域以及家庭、商场和车载照明领域,未来将向大尺寸显示产品应用领域扩展。
有机电致发光的过程包括载流子经外加电场作用,电子、空穴分别从阴极、阳极注入;电子和空穴分别从电子传输层和空穴传输层向发光层迁移;电子和空穴在发光层复合,产生激子,跃迁至激发态;处于激发态的激子迁移,将能量传递给发光材料;电致发光,发光分子从激发态回到基态,激发能量以光能形式释放。传统荧光材料激子利用率低,理论最高利用仅为25%的单线态激子;磷光材料使用重金属铱等材料,成本高昂。因此,高效利用三线态激子,是实现效率突破的关键。2012年,adachi等通过结构设计,提出热激活延迟荧光(tadf)机理,将75%的三线态激子转变成单线态激子,实现100%的内量子效率,是oled领域的一大进步。
tadf类材料有一个明显缺陷就是发光光谱过宽,发光颜色不纯。多重共振诱导的热激活延迟荧光(mr-tadf)材料可有效解决此问题。通过分子内共振效应,实现homo/lumo分离同时,最大限度减少长程的电荷转移态,实现高效率和窄带发光。但mr-tadf类材料发光颜色难以调控,其发光局限在蓝光-深蓝光区域,同时伴随严重的器件效率滚降。近年来,绿光mr-tadf材料有所发展,但有关红光的mr-tadf材料的报道仍是寥寥无几。目前,仅清华段炼课题组(10.26434/chemrxiv.13202975.v1)和日本takumayasuda(j.am.chem.soc.2020,142,46,19468–19472)报道两类红光硼氮化合物,但其荧光发射光谱峰的半峰宽较宽,这很大程度上限制了mr-tadf材料在全彩显示及照明领域的进一步应用。
因此,现有技术仍有待于改进和发展。
技术实现要素:
鉴于上述现有技术的不足,本发明的目的在于提供一种热激活延迟红光荧光材料、有机电致发光器件与显示装置,旨在解决现有荧光材料作为有机电致发光器件的发光材料,其色纯度低和发光效率低的问题。
本发明的技术方案如下:
一种热激活延迟红光荧光材料,其通式如下式(1)所示:
式(1)中,x1、x2、x3、x4分别独立地选自nr、o、s、asr或bir,所述nr、asr和bir中的r分别独立地选自氢、氘、烷基、单环芳烃、稠环芳烃、单环杂芳烃或稠环杂芳烃;环a、环c、环d、环e分别独立地选自五元环、六元环或七元环;ra、rc、rd、re分别独立地选自氢、氘、卤素、氰基、取代或未取代的烷基或芳香基团。
优选地,所述热激活延迟红光荧光材料的结构选自下式所示中的一种:
式(2-1)至(2-2)中,x1、x2、x3、x4分别独立地选自n、as或bi,r1-r28分别独立地选自氢、氘、卤素、氰基、取代或未取代的烷基、烯基、烷氧基或硫代烷氧基、单环芳烃或稠环芳烃、单环杂芳烃或稠环杂芳烃。
优选地,所述热激活延迟红光荧光材料的结构如下式(2-3)所示:
式(2-3)中,x1、x2、x3、x4分别独立地选自n、as或bi,r1-r28分别独立地选自氢、氘、卤素、氰基、取代或未取代的烷基、烯基、烷氧基或硫代烷氧基、单环芳烃或稠环芳烃、单环杂芳烃或稠环杂芳烃;x5、x6、x7、x8分别独立地选自单键、o、s或cra,其中ra分别独立地选自取代或未取代的烷基、单环芳烃或稠环芳烃、单环杂芳烃或稠环杂芳烃。
优选地,所述热激活延迟红光荧光材料的结构选自下式所示中的一种:
式(2-4)至(2-6)中,x分别独立地选自o或s;r1-r20分别独立地选自氢、氘、卤素、氰基、取代或未取代的烷基、烯基、烷氧基或硫代烷氧基、单环芳烃或稠环芳烃、单环杂芳烃或稠环杂芳烃。
优选地,所述热激活延迟红光荧光材料的结构如下式所示:
式(2-7)中,x分别独立地选自o或s;r1-r36分别独立地选自氢、氘、卤素、氰基、取代或未取代的烷基、烯基、烷氧基或硫代烷氧基、单环芳烃或稠环芳烃、单环杂芳烃或稠环杂芳烃。
优选地,所述热激活延迟红光荧光材料的结构如下式(2-8)所示:
式(2-8)中,x分别独立地选自o、s或se。
优选地,所述热激活延迟红光荧光材料的结构如下式(2-9)所示:
式(2-9)中,r1-r44分别独立地选自氢、氘、卤素、氰基、取代或未取代的烷基、烯基、烷氧基或硫代烷氧基、单环芳烃或稠环芳烃、单环杂芳烃或稠环杂芳烃。
所述热激活延迟红光荧光材料的结构选自下式所示中的一种:
一种有机电致发光器件,包括发光层,所述发光层包括本发明所述的热激活延迟红光荧光材料。
本发明中所述发光层包括主体材料和客体材料,所述主体材料可以为mcp、mcbp、cbp等,但不限于此;所述客体材料为本发明所述的热激活延迟红光荧光材料。
本发明的有机电致发光器件的器件结构优选为:基板/阳极/空穴传输层/发光层/电子传输层/阴极、基板/阳极/空穴传输层/发光层/电子传输层/电子注入层/阴极、基板/阳极/空穴注入层/空穴传输层/发光层/电子传输层/电子注入层/阴极、基板/阳极/空穴注入层/空穴传输层/激子阻挡层/发光层/电子传输层/电子注入层/阴极等,当然有机电致发光器件的结构不限于此。
一种显示装置,包括本发明所述的有机电致发光器件。
下面对本发明的如上通式的热激活延迟红光荧光材料的制备方法进行介绍:
其中,y1-y4分别独立地选自f、cl、br、i;y5、y6分别独立地选自h、f、cl、br、i;x选自n、as、bi中的一种。本发明中制备方法主要流程:首先,利用c-x偶联反应制备中间体;继而,添加正丁基锂或叔丁基锂进行邻位金属化;接着,添加三溴化硼等,进行锂-硼或锂-磷的金属交换后,添加n,n-二异丙基乙胺等布朗斯特碱,进行串联式硼杂弗里德-克拉夫茨反应,得到目标物。
有益效果:本发明提供的如上结构通式的化合物具有热激活延迟荧光特性,包含b-π-b线性结构;同时引入杂原子结构基元、重原子、七元环等,使其荧光发射峰红移,荧光发射光谱峰的半峰全宽仅有20-40nm,从而获得红光/深红光的热激活延迟荧光材料;同时,引入氘元素,提升器件寿命稳定性。本发明如上结构通式的化合物作为有机电致发光器件的发光材料,具有发光效率高、色纯度高和效率滚降小等优点。
附图说明
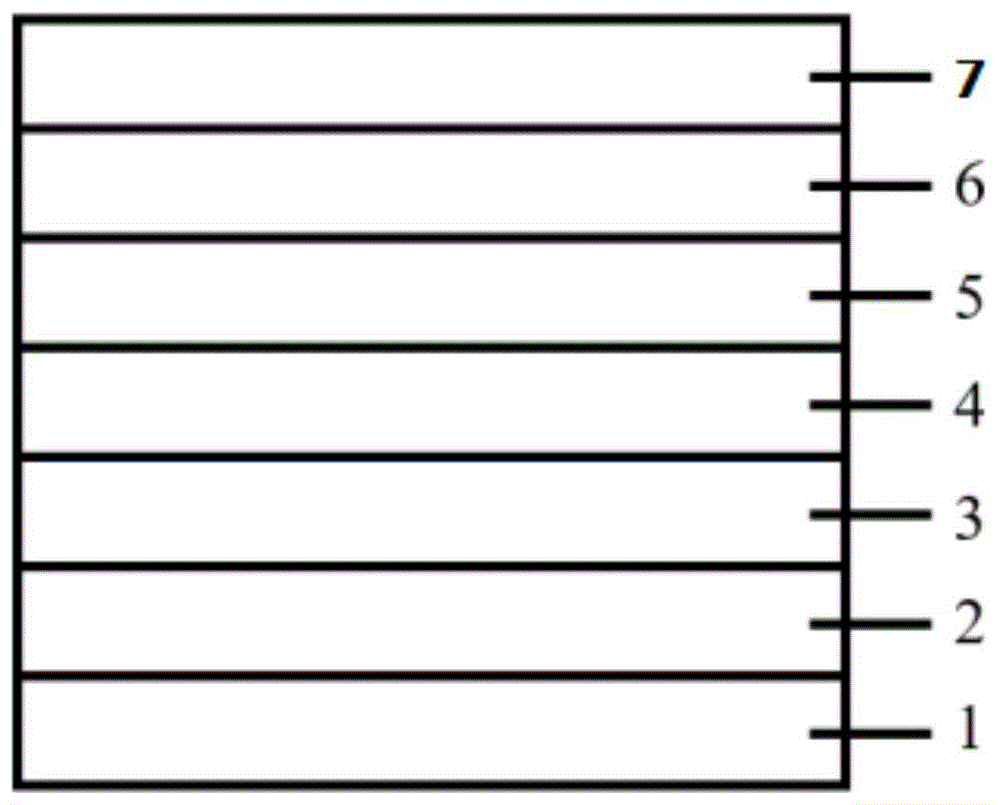
图1为本发明所制备的有机电致发光器件的结构示意图,图中,1为基板,2为阳极,3为空穴传输层,4为有机发光层,5为电子传输层,6为阴极。
具体实施方式
本发明提供一种热激活延迟红光荧光材料、有机电致发光器件与显示装置,为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
以下通过具体实施例对本发明的热激活延迟红光荧光材料的制备方法作进一步的介绍。
实施例1:化合物1-f的合成
在圆底烧瓶中,将1-a(1.0eq)、1-b(1.0eq)、pd2(dba)3(0.045eq)、三叔丁基膦(0.025eq)和叔丁醇钠(2.0eq)加入到甲苯,升温到100℃进行反应。反应结束后,加入水和二氯甲烷,收集有机相,真空旋干溶剂,过柱子得化合物1-c。500ml圆底烧瓶中,将1-c(4.4eq)和碳酸铯(12.0eq)加入到n,n-二甲基甲酰胺dmf中,室温搅拌30分钟;然后,加入化合物1-d(1.0eq),升温至150℃,反应12小时。反应结束后,加入水稀释,抽滤得固体,接着在热乙醇中回流4小时,过滤得固体1-e。氮气氛围下,将正丁基锂的正己烷溶液(3.0eq)缓慢加入到0℃的前驱体1-e(1.0eq)的叔丁苯溶液中,搅拌0.5小时,然后升温至60℃,反应2小时。反应结束后降温至-40℃,缓慢加入三溴化硼(3.6eq),室温继续搅拌0.5小时。0℃下加入n,n-二异丙基乙胺(5.0eq),并在120℃下继续反应5小时后停止。等反应体系降温到室温,加入乙酸钠水溶液,并用二氯甲烷萃取,真空旋干溶剂,过硅胶柱,得目标化合物1-f。
实施例2:化合物2-f的合成
500ml圆底烧瓶中,将3,6-二叔丁基咔唑2-b(4.4eq)和碳酸铯(12.0eq)加入到n,n-二甲基甲酰胺dmf中,室温搅拌30分钟;然后,加入化合物2-a(1.0eq),升温至150℃,反应12小时。反应结束后,加入水稀释,抽滤得固体,接着在热乙醇中回流4小时,过滤得固体2-c。氮气氛围下,将化合物2-c(1.0eq)、二苯胺(2.2eq)、pd2(dba)3(0.06eq)、三叔丁基膦(0.10eq)和叔丁醇钠(3.3eq)加入到干燥的甲苯中,升温到110℃,反应过夜。反应结束后,冷却到室温,加入水和乙酸乙酯,收集有机相,加入无水硫酸镁干燥,真空旋干溶剂,过硅胶柱,得产物2-e。氮气氛围下,将正丁基锂的正己烷溶液(3.0eq)缓慢加入到0℃的前驱体2-e(1.0eq)的叔丁苯溶液中,搅拌0.5小时,然后升温至60℃,反应2小时。反应结束后降温至-40℃,缓慢加入三溴化硼(3.6eq),室温继续搅拌0.5小时。0℃下加入n,n-二异丙基乙胺(5.0eq),并在120℃下继续反应5小时后停止。反应体系恢复到室温,加入乙酸钠水溶液,并用二氯甲烷萃取,真空旋干溶剂,过硅胶柱,得目标化合物2-f。
实施例3:化合物3-d的合成
500ml圆底烧瓶中,将吩恶嗪3-b(4.4eq)和碳酸铯(12.0eq)加入到n,n-二甲基甲酰胺dmf中,室温搅拌30分钟;然后,加入化合物3-a(1.0eq),升温至150℃,反应12小时。反应结束后,加入水稀释,抽滤得固体,接着在热乙醇中回流4小时,过滤得固体3-c。氮气氛围下,将正丁基锂的正己烷溶液(3.0eq)缓慢加入到0℃的前驱体3-c(1.0eq)的叔丁苯溶液中,搅拌0.5小时,然后升温至60℃,反应2小时。反应结束后降温至-40℃,缓慢加入三溴化硼(3.6eq),室温继续搅拌0.5小时。0℃下加入n,n-二异丙基乙胺(5.0eq),并在120℃下继续反应5小时后停止。反应体系恢复到室温,加入乙酸钠水溶液,并用二氯甲烷萃取,真空旋干溶剂,过硅胶柱,得目标化合物3-d。
实施例4:化合物4-f的合成
在圆底烧瓶中,将4-a(1.0eq)、4-b(1.0eq)、pd2(dba)3(0.045eq)、三叔丁基膦(0.025eq)和叔丁醇钠(2.0eq)加入到甲苯,升温到100℃进行反应。反应结束后,加入水和二氯甲烷,收集有机相,真空旋干溶剂,过柱子得化合物4-c。500ml圆底烧瓶中,将4-c(4.4eq)和碳酸铯(12.0eq)加入到n,n-二甲基甲酰胺dmf中,室温搅拌30分钟;然后,加入化合物4-d(1.0eq),升温至150℃,反应12小时。反应结束后,加入水稀释,抽滤得固体,接着在热乙醇中回流4小时,过滤得固体4-e。氮气氛围下,将正丁基锂的正己烷溶液(3.0eq)缓慢加入到0℃的前驱体4-e(1.0eq)的叔丁苯溶液中,搅拌0.5小时,然后升温至60℃,反应2小时。反应结束后降温至-40℃,缓慢加入三溴化硼(3.6eq),室温继续搅拌0.5小时。0℃下加入n,n-二异丙基乙胺(5.0eq),并在120℃下继续反应5小时后停止。等反应体系降温到室温,加入乙酸钠水溶液,并用二氯甲烷萃取,真空旋干溶剂,过硅胶柱,得目标化合物4-f。
实施例5:化合物5-f的合成
在圆底烧瓶中,将5-a(1.0eq)、5-b(1.0eq)、pd2(dba)3(0.045eq)、三叔丁基膦(0.025eq)和叔丁醇钠(2.0eq)加入到甲苯,升温到100℃进行反应。反应结束后,加入水和二氯甲烷,收集有机相,真空旋干溶剂,过柱子得化合物5-c。500ml圆底烧瓶中,将5-c(4.4eq)和碳酸铯(12.0eq)加入到n,n-二甲基甲酰胺dmf中,室温搅拌30分钟;然后,加入化合物5-d(1.0eq),升温至150℃,反应12小时。反应结束后,加入水稀释,抽滤得固体,接着在热乙醇中回流4小时,过滤得固体5-e。氮气氛围下,将正丁基锂的正己烷溶液(3.0eq)缓慢加入到0℃的前驱体5-e(1.0eq)的叔丁苯溶液中,搅拌0.5小时,然后升温至60℃,反应2小时。反应结束后降温至-40℃,缓慢加入三溴化硼(3.6eq),室温继续搅拌0.5小时。0℃下加入n,n-二异丙基乙胺(5.0eq),并在120℃下继续反应5小时后停止。等反应体系降温到室温,加入乙酸钠水溶液,并用二氯甲烷萃取,真空旋干溶剂,过硅胶柱,得目标化合物5-f。
实施例6:化合物6-d的合成
500ml圆底烧瓶中,将6-b(4.4eq)和碳酸铯(12.0eq)加入到n,n-二甲基甲酰胺dmf中,室温搅拌30分钟;然后,加入化合物6-a(1.0eq),升温至150℃,反应12小时。反应结束后,加入水稀释,抽滤得固体,接着在热乙醇中回流4小时,过滤得固体6-c。氮气氛围下,将正丁基锂的正己烷溶液(3.0eq)缓慢加入到0℃的前驱体6-c(1.0eq)的叔丁苯溶液中,搅拌0.5小时,然后升温至60℃,反应2小时。反应结束后降温至-40℃,缓慢加入三溴化硼(3.6eq),室温继续搅拌0.5小时。0℃下加入n,n-二异丙基乙胺(5.0eq),并在120℃下继续反应5小时后停止。反应体系恢复到室温,加入乙酸钠水溶液,并用二氯甲烷萃取,真空旋干溶剂,过硅胶柱,得目标化合物6-d。
实施例7:化合物7-d的合成
500ml圆底烧瓶中,将化合物7-b(4.4eq)和碳酸铯(12.0eq)加入到n,n-二甲基甲酰胺dmf中,室温搅拌30分钟;然后,加入化合物7-a(1.0eq),升温至150℃,反应12小时。反应结束后,加入水稀释,抽滤得固体,接着在热乙醇中回流4小时,过滤得固体7-c。氮气氛围下,将正丁基锂的正己烷溶液(3.0eq)缓慢加入到0℃的前驱体7-c(1.0eq)的叔丁苯溶液中,搅拌0.5小时,然后升温至60℃,反应2小时。反应结束后降温至-40℃,缓慢加入三溴化硼(3.6eq),室温继续搅拌0.5小时。0℃下加入n,n-二异丙基乙胺(5.0eq),并在120℃下继续反应5小时后停止。反应体系恢复到室温,加入乙酸钠水溶液,并用二氯甲烷萃取,真空旋干溶剂,过硅胶柱,得目标化合物7-d。
实施例8:化合物8-f的合成
在圆底烧瓶中,将8-a(1.0eq)、8-b(1.0eq)、pd2(dba)3(0.045eq)、三叔丁基膦(0.025eq)和叔丁醇钠(2.0eq)加入到甲苯,升温到100℃进行反应。反应结束后,加入水和二氯甲烷,收集有机相,真空旋干溶剂,过柱子得化合物8-c。500ml圆底烧瓶中,将8-c(4.4eq)和碳酸铯(12.0eq)加入到n,n-二甲基甲酰胺dmf中,室温搅拌30分钟;然后,加入化合物8-d(1.0eq),升温至150℃,反应12小时。反应结束后,加入水稀释,抽滤得固体,接着在热乙醇中回流4小时,过滤得固体8-e。氮气氛围下,将正丁基锂的正己烷溶液(3.0eq)缓慢加入到0℃的前驱体8-e(1.0eq)的叔丁苯溶液中,搅拌0.5小时,然后升温至60℃,反应2小时。反应结束后降温至-40℃,缓慢加入三溴化硼(3.6eq),室温继续搅拌0.5小时。0℃下加入n,n-二异丙基乙胺(5.0eq),并在120℃下继续反应5小时后停止。等反应体系降温到室温,加入乙酸钠水溶液,并用二氯甲烷萃取,真空旋干溶剂,过硅胶柱,得目标化合物8-f。
实施例9:化合物9-f的合成
在圆底烧瓶中,将9-a(1.0eq)、9-b(1.0eq)、pd2(dba)3(0.045eq)、三叔丁基膦(0.025eq)和叔丁醇钠(2.0eq)加入到甲苯,升温到100℃进行反应。反应结束后,加入水和二氯甲烷,收集有机相,真空旋干溶剂,过柱子得化合物9-c。500ml圆底烧瓶中,将9-c(4.4eq)和碳酸铯(12.0eq)加入到n,n-二甲基甲酰胺dmf中,室温搅拌30分钟;然后,加入化合物9-d(1.0eq),升温至150℃,反应12小时。反应结束后,加入水稀释,抽滤得固体,接着在热乙醇中回流4小时,过滤得固体9-e。氮气氛围下,将正丁基锂的正己烷溶液(3.0eq)缓慢加入到0℃的前驱体9-e(1.0eq)的叔丁苯溶液中,搅拌0.5小时,然后升温至60℃,反应2小时。反应结束后降温至-40℃,缓慢加入三溴化硼(3.6eq),室温继续搅拌0.5小时。0℃下加入n,n-二异丙基乙胺(5.0eq),并在120℃下继续反应5小时后停止。等反应体系降温到室温,加入乙酸钠水溶液,并用二氯甲烷萃取,真空旋干溶剂,过硅胶柱,得目标化合物9-f。
以下通过具体实施例对本发明的有机电致发光器件作进一步的介绍。
如下实施例1-6分别提供一种有机电致发光器件,如图1所示,其器件结构均依次包括ito阳极1、空穴注入层(hil)2、空穴传输层(htl)3、发光层(eml)4、电子传输层(etl)5、电子注入层(eil)6和阴极7。
实施例1
本实施例的器件结构如下所示:ito/hi(10nm)/ht(50nm)/host:3wt%化合物1-f(30nm)/et(30nm)/liq(1nm)/al(100nm)
其中,空穴注入层的材料为hi,一般总厚度为5-20nm,本实施例为10nm;空穴传输层的材料为ht,总厚度一般为5-100nm,本实施例为50nm;host为有机发光层宽带隙的主体材料,化合物1-f为客体材料且掺杂浓度为3wt%,有机发光层厚度一般为1-100nm,本实施例为30nm;电子传输层的材料为et,总厚度一般为5-100nm,本实施例为30nm;电子注入层及阴极材料为liq(1nm)和金属铝(100nm)。
针对本实施例制备得到的有机电致发光器件d1测定器件性能结果如下:施加直流电压,可获得波长683nm、半峰宽35nm、cie色坐标(x,y)=(0.70,0.29)、外量子效率eqe为26%的红色发光。
实施例2
与实施例1的制备方法相同,区别在于,将发光层客体材料替换为化合物3-d。器件结构如下:ito/hi(10nm)/ht(50nm)/host:3wt%化合物3-d(30nm)/et(30nm)/liq(1nm)/al(100nm)
针对本实施例制备得到的有机电致发光器件d2测定器件性能结果如下:施加直流电压,可获得波长657nm、半峰宽35nm、cie色坐标(x,y)=(0.70,0.30)、外量子效率eqe为25%的红色发光。
实施例3
与实施例1的制备方法相同,区别在于,将发光层客体材料替换为化合物4-f。器件结构如下:ito/hi(10nm)/ht(50nm)/host:3wt%化合物4-f(30nm)/et(30nm)/liq(1nm)/al(100nm)
针对本实施例制备得到的有机电致发光器件d3测定器件性能结果如下:施加直流电压,可获得波长680nm、半峰宽28nm、cie色坐标(x,y)=(0.69,0.32)、外量子效率eqe为26.7%的红色发光。
实施例4
与实施例1的制备方法相同,区别在于,将发光层客体材料替换为化合物5-f。器件结构如下:ito/hi(10nm)/ht(50nm)/host:3wt%化合物5-f(30nm)/et(30nm)/liq(1nm)/al(100nm)
针对本实施例制备得到的有机电致发光器件d4测定器件性能结果如下:施加直流电压,可获得波长677nm、半峰宽33nm、cie色坐标(x,y)=(0.70,0.28)、外量子效率eqe为22.1%的红色发光。
实施例5
与实施例1的制备方法相同,区别在于,将发光层客体材料替换为化合物6-d。器件结构如下:ito/hi(10nm)/ht(50nm)/host:3wt%化合物6-d(30nm)/et(30nm)/liq(1nm)/al(100nm)
针对本实施例制备得到的有机电致发光器件d6测定器件性能结果如下:施加直流电压,可获得波长634nm、半峰宽30nm、cie色坐标(x,y)=(0.66,0.31)、外量子效率eqe为26.3%的红色发光。
实施例6
与实施例1的制备方法相同,区别在于,将发光层客体材料替换为化合物10-f。器件结构如下:ito/hi(10nm)/ht(50nm)/host:3wt%化合物10-f(30nm)/et(30nm)/liq(1nm)/al(100nm)
针对本实施例制备得到的有机电致发光器件d6测定器件性能结果如下:施加直流电压,可获得波长681nm、半峰宽26nm、cie色坐标(x,y)=(0.71,0.30)、外量子效率eqe为26.3%的红色发光。
上述各个实施例中所采用的各类有机材料的结构式如下:
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!