一种表达单元、重组慢病毒表达载体、重组慢病毒及其制备方法和应用
peptide(cgrp)和p物质(substance p)。这些外周痛觉信号最终通过drg和tgn神经元传递到中枢神经系统(cns)和大脑。周围伤害性刺激drg小直径无髓鞘c和薄髓鞘aδ感觉纤维,感觉纤维的异常兴奋可增强因有害刺激而诱导的痛觉过敏与无害的刺激而诱导的痛觉超敏,进而引起局部多种痛觉神经递质(如谷氨酸、p物质和cgrp等神经多肽)释放。这些神经递质可以调节固有免疫细胞和获得性免疫细胞(巨噬细胞、内皮细胞、肥大细胞、滑膜细胞及t细胞等)的功能,亦可增加促炎性细胞因子例如:肿瘤坏死因子-α(tnf-α)、白细胞介素(interleukin)1(il-1)、il-6和il-17等、前列腺素e2及胶原酶的产生和释放,加剧外周部位的炎症反应。
5.drg和tgn神经元中p物质和cgrp等神经肽的分泌需要三种snare(可溶性n-乙基马来酰亚胺敏感因子附着蛋白受体):突触小泡相关膜蛋白亚型1(vesicle associated membrane protein 1,vamp1)、25kda突触相关蛋白(synaptosome associatedprotein-25,snap-25)和突触融合蛋白-1(syntaxin-1),三者形成snare复合体介导大致密核心颗粒(large dense core vesicle,ldcv)与胞膜融合和神经肽的分泌。此外,tnf-α可上调和活化神经元膜上瞬时感受器电位通道香草素受体1(trpv1)与瞬时感受器电位a1通道(trpa1),用肉毒杆菌毒素(botulinum neurotoxin,bont,简称肉毒素)的蛋白酶切割snare,不仅抑制了神经肽的分泌,而且阻止了tnf-α引起的trpv1和trpa1在神经膜上的过表达。因此,肉毒素的蛋白酶具有抑制痛觉神经递质过度释放和恢复痛觉传导离子通道蛋白正常表达治疗慢性疼痛的生物活性。
6.肉毒素是由厌氧肉毒梭菌(clostridium botulinum)产生的一种外毒素。肉毒素由一个约50kda的蛋白酶轻链(lc)和一个约100kda的重链(hc)组成,通过一个非共价二硫键连接。肉毒素a型和b型已被应用在治疗肌肉痉挛和其他肌肉运动紊乱症,包括癫痫,肌张力障碍,斜视等治疗中。肉毒素作用时效长,一次注射治疗效果可达3-6个月。在美容界同样受到人们的关注,同时,它还能缓解部分偏头痛病人和骨关节炎病人的疼痛。自然界已分离7种同源型肉毒素(bont/a-g),通过生物信息学发现了第8种新型肉毒素x。每种bont对snare蛋白底物均具有不同的特异性切割位点。例如:snap-25被发现被bont/a、bont/c1和bont/e切割;vamp-1,2,3被bont/b、bont/d、bont/f、bont/g切割;破伤风毒素也可切割vamp-1,2和3。syntaxin 1可被bont/c1切割。bont的镇痛原理就在于bont对snare蛋白切割,从而抑制痛觉递质过度分泌。反之,也证明了snare蛋白在痛觉传导过程中的重要作用。
7.目前临床上,用于治疗疼痛的非甾体类解热镇痛药或阿片类麻醉性镇痛药物具有作用时间短且毒副作用大的特点。近五年在全球范围内,治疗神经病理性疼痛常用药物普瑞巴林年均销售额高达50亿美元,但治疗效果不尽人意。慢性疼痛患者病程较长,病因复杂,在使用传统药物过程中易出现耐药性,并对其他器官产生副作用,显然对于慢性疼痛患者这类药物并不适用,因此慢性疼痛病人急切需要一种长效的药物镇痛,以缓解心理,生理的不悦感受。
技术实现要素:
8.有鉴于此,本发明的目的在于提供一种表达单元、重组慢病毒表达载体、重组慢病毒及其制备方法和应用,能够有效切割snare蛋白,抑制痛觉神经递质的分泌,达到基因治疗疼痛的目的。
9.为解决上述技术问题,本发明提供了以下技术方案:
10.一种表达单元,所述表达单元包括启动子和外源基因;所述启动子为人源pirt基因启动子或rft-i启动子。
11.优选的,所述外源基因为肉毒素蛋白酶基因或破伤风毒素蛋白酶基因。
12.优选的,所述人源pirt基因启动子的核苷酸序列如seq id no.1所示,所述rft-i启动子的核苷酸序列如seq id no.2所示。
13.本发明提供了一种重组慢病毒表达载体,所述重组慢病毒表达载体以慢病毒骨架载体syn-dsred-syn-gfp为基础,在多克隆位点插入上述的表达单元。
14.优选的,所述多克隆位点为nsi i和ecor i的酶切位点。
15.本发明提供了上述重组慢病毒表达载体的构建方法,包括以下步骤:
16.1)将人源pirt基因启动子插入到syn-dsred-syn-gfp慢病毒骨架载体的nsi i与nhe i酶切位点之间,得到syn-dsred-hpirt-gfp载体;
17.2)将外源基因插入到syn-dsred-hpirt-gfp载体的nhe i和ecor i酶切位点之间,即得含hpirt-外源基因表达单元的重组慢病毒表达载体;
18.3)将rft-i启动子插入到上述(2)重组慢病毒表达载体的nsi i与nhe i酶切位点之间,即得含rft-i-外源基因表达单元的重组慢病毒表达载体。
19.本发明还提供了包括上述重组慢病毒表达载体的重组慢病毒。
20.本发明还提供了上述重组慢病毒的制备方法,具体包括以下步骤:
21.将上述的重组慢病毒表达载体与慢病毒包膜载体及包装载体共转染细胞系即得。
22.优选的,所述慢病毒包膜载体为pmd2.g;所述包装载体为pspax2;所述细胞系为293t细胞。
23.本发明还提供了上述表达单元、重组慢病毒表达载体或重组慢病毒在制备治疗慢性疼痛药物中的应用。
24.本发明提供了一种表达单元、重组慢病毒表达载体和重组慢病毒,慢病毒感染培养的背根神经节和三叉神经节神经元,有效切割了snare蛋白,抑制了痛觉神经递质(p物质)的分泌,具备特异性抑制外周痛觉发生和信号传导的特点,从而实现基因治疗疼痛的目的。
附图说明
25.图1构建的两种重组慢病毒表达载体常用限制性内切酶图谱;
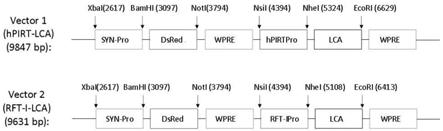
26.图2构建的两种重组慢病毒表达载体酶切核酸电泳结果;
27.图3 hpirt-lca病毒感染小鼠drg和tgn神经元细胞切割snap-25的结果;
28.图4 rft-i-lca病毒感染小鼠drg和tgn神经元细胞切割snap-25的结果;
29.图5 hpirt-lca病毒感染小鼠drg,tgn神经元细胞抑制p物质分泌的效果图;
30.图6验证hpirt-lca病毒感染小鼠drg神经元细胞具有切割snap-25的长效性的效果图。
具体实施方式
31.本发明提供了一种表达单元,所述表达单元包括启动子和外源基因;所述启动子
为人源pirt基因启动子或rft-i启动子。
32.本发明中,所述人源pirt基因为与磷脂酰肌醇互作的瞬时受体电位通道的调节蛋白基因(phosphoinositide-interacting regulator of transient receptorpotential channels),所述人源pirt基因启动子的核苷酸序列是由pirt基因转录起始位点上游和下游共928bp的核苷酸序列构成。本发明中,所述人源pirt基因启动子优选为人工合成;所述人工合成的人源pirt基因启动子的核苷酸序列如seq id no.1所示;其中,5
′
端插入nsi i限制性内切酶序列atgcat,3
′
端插nhe i限制性内切酶序列gctagc。本发明中,采用pirt启动子是为了限制插入的肉毒素蛋白酶外源基因在drg和tgn外周神经中特异表达,在脊髓和脑中不表达,提高药物的安全性。
33.本发明中,所述rft-i为兔源β-半乳糖苷α1,2-岩藻糖基转移酶基因(rabbitβ-galactosideα1,2-fucosyltransferase gene),所述rft-i启动子为rft-i最小启动子(hitoshi s等作者发表于j biol chem.1999;274(1):389-396)。本发明中,所述rft-i启动子优选为人工合成;所述人工合成的rft-i启动子的核苷酸序列如seq id no.2所示;其中,5
′
端插入nsi i限制性内切酶序列atgcat,3
′
端插nhe i限制性内切酶序列gctagc。本发明中,利用rft-i启动子背根神经节神经元特异表达外源基因的生物活性,在其下游插入肉毒素蛋白酶外源基因,从而限制插入的肉毒素蛋白酶外源基因主要在drg和tgn中表达,在其他细胞与组织中不表达或微量表达。
34.本发明中,所述外源基因优选为肉毒素蛋白酶基因或破伤风毒素蛋白酶基因。本发明中,所述肉毒素蛋白酶基因或破伤风毒素蛋白酶基因优选的在体外合成;所述肉毒素蛋白酶基因的核苷酸序列如seq id no.3所示;其中,5
′
端插入nhe i限制性内切酶序列gctagc,3
′
端插eco ri限制性内切酶序列gaattc。
35.本发明提供了一种重组慢病毒表达载体,所述重组慢病毒表达载体以慢病毒骨架载体syn-dsred-syn-gfp为基础,在多克隆位点插入上述的表达单元。本发明中,对所述慢病毒骨架载体syn-dsred-syn-gfp的来源并没有特殊限定,本发明优选的采用sergio gasc
ó
n等作者于2008年发表在j neurosci methods.168(1):104-112中的慢病毒骨架载体。所述慢病毒骨架载体含有两个突触蛋白1(synapsin 1)基因启动子(简写为syn);其中,第一个突触蛋白1基因启动子用于表达dsred报道基因,第二个突触蛋白1基因启动子用于表达gfp报道基因。本发明中,所述多克隆位点为nsi i和ecor i的酶切位点。
36.本发明提供了上述重组慢病毒表达载体的构建方法,包括以下步骤:
37.1)将人源pirt基因启动子插入到syn-dsred-syn-gfp慢病毒骨架载体的nsi i与nhe i酶切位点之间,得到syn-dsred-hpirt-gfp载体;
38.2)将外源基因插入到syn-dsred-hpirt-gfp载体的nhe i和ecor i酶切位点之间,即得含hpirt-外源基因表达单元的重组慢病毒表达载体;
39.3)将rft-i启动子插入到上述(2)重组慢病毒表达载体的nsi i与nhe i酶切位点之间,即得含rft-i-外源基因表达单元的重组慢病毒表达载体。
40.本发明中,所述人源pirt基因启动子或rft-i启动子优选的在体外进行合成。本发明对所述启动子插入到syn-dsred-syn-gfp慢病毒骨架载体的方法并没有特殊限定,采用本领域常规的分子生物学基本方法即可。
41.本发明还提供了包括上述重组慢病毒表达载体的重组慢病毒。本发明中,所述慢
病毒同时含有人的突触蛋白1(synapsin 1)基因启动子,该启动子为神经细胞特异性。本发明优选的表达载体在突触蛋白1基因启动子的下游插入有报告荧光基因dsred。所述dsred在细胞中表达可作为病毒感染和转导外源基因效果的标记。
42.本发明还提供了上述重组慢病毒的制备方法,具体包括以下步骤:
43.将上述的重组慢病毒表达载体与慢病毒包膜载体及包装载体共转染细胞系即得。
44.本发明中,所述慢病毒包膜载体优选为pmd2.g;所述包装载体优选为pspax2;所述细胞系优选为293t细胞。本发明对所述转染的方法并没有特殊限定,采用本领域常规的转染方法即可。
45.本发明还提供了上述表达单元、重组慢病毒表达载体或重组慢病毒在制备治疗慢性疼痛药物中的应用。
46.为使本发明的目的、技术方案和优点更加清楚明白,下面结合实施例对本发明进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
47.下述实施例中,如无特殊说明,均为常规方法。
48.下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
49.实施例1
50.1、构建重组慢病毒表达载体1
51.(1)利用ensembl工具分析人pirt基因,人工合成pirt基因启动子和5
′
端限制性内切酶nsi i及3
′
端nhei的序列seq id no.1;
52.(2)将含有步骤(1)人工合成的pirt基因启动子元件的质粒用nsii和nhei双酶切,凝胶电泳回收约930bp,即得hpirt启动子片段。
53.(3)将hpirt启动子片段采用t4dna连接酶与syn-dsred-syn-gfp质粒预先用nsi i和nhe i双酶切并用凝胶电泳回收的大片段连接,转化top 10感受态细菌,挑取单菌落进行细菌培养并抽提质粒进行nsi i和nhe i酶切及测序鉴定,得到syn-dsred-hpirt-gfp载体;
54.(4)体外合成序列如seq id no.3的肉毒素a型蛋白酶基因,将其插入到syn-dsred-hpirt-gfp载体的nhe i和ecor i酶切位点之间取代gfp基因,得到syn-dsred-hpirt-lca载体,即为重组慢病毒表达载体1,简称为hpirt-lca,具体如附图1所示。
55.所述的hpirt-lca利用人的pirt启动子来表达lca基因,同时可以通过synapsin 1启动子在神经细胞中表达报告基因dsred。
56.2、构建重组慢病毒表达载体2
57.人工合成rft-i最小启动子序列seq id no.2,采用上述相同的分子生物学方法将其插入到syn-dsred-hpirt-lca载体的nsi i和nhe i位点之间,替换其中的hpirt基因启动子,从而得到syn-dsred-rft-i-lca载体,即为重组慢病毒表达载体2,简称为rft-i-lca,具体如附图1所示。
58.所述的rft-i-lca利用rft-i启动子来表达lca基因,同时可以通过synapsin 1启动子在神经细胞中表达报告基因dsred。
59.实施例2重组慢病毒表达载体的鉴定与提取
60.1、分组双酶切验证重组慢病毒表达载体构建的正确性
61.取200μl离心管,向离心管中依次加入表1中的每种试剂,轻轻吸打混匀,置于37℃水浴锅中反应3h。3h后向每个离心管中加入10μl 6
×
loading buffer终止酶切轻轻吹打混
匀。制备0.7%琼脂糖核酸胶,上样进行琼脂糖凝胶电泳。电泳完毕将核酸胶置于g-box中拍照,得到附图2。
62.表1加入试剂
63.试剂体积(μl)重组慢病毒表达载体dna(1μg/μl)110
×
neb buffer5h2o42酶11酶21total50
64.其中,鉴定不同的启动子和下游的表达基因,添加的酶1和酶2组合并不相同,具体如下:
65.验证插入的hpirt或rft-i启动子,酶1为nsi i酶,酶2为nhe i酶;
66.验证插入的lca基因,酶1为nhe i酶,酶2为ecor i酶;
67.鉴定骨架载体synapsin 1启动子,酶1为xba i酶,酶2为bamh i酶:
68.鉴定骨架载体dsred报道基因,酶1为bamh i酶,酶2为not i酶。
69.由附图2可以看出,重组慢病毒表达载体1(hpirt-lca)用nsi i+nhe i双酶切得到两条带,其中较小的带大小约930bp,与hpirtpro大小相符。nhe i+ecor i双酶切也得到两条带,其中较小的条带大小约1305bp,与肉毒素a型蛋白酶lca大小相符。xba i+bamh i和bamh i+not i双酶切分别得到两条小条带为synapsin 1启动子(480bp)和dsred(697bp),均与预期骨架慢病毒表达载体相符。
70.重组慢病毒表达载体2(rft-i-lca)用nsi i+nhe i双酶切得到两条带,其中较小的条带大小约715bp,与rft-i pro大小相符。nhe i+ecor i双酶切也得到两条带,其中较小的条带大小约1305bp,与肉毒素a型蛋白酶lca大小相符。xba i+bamh i和bamh i+not i双酶切分别得到两条小条带为synapsin 1启动子(480bp)和dsred(697bp),均与预期骨架慢病毒表达载体相符。
71.2、重组慢病毒表达载体的转化
72.将1μl重组慢病毒表达载体加入至50μl top 10感受态细菌中,使二者混合,冰上孵育30min,42℃水浴60s,再置于冰上孵育5min,加入400μl lb培养基后放置于37℃,220rpm摇床培养45min。取50μl菌液涂布氨苄抗性培养板,涂布均匀后,放置37℃培养箱中培养18h。
73.3、重组慢病毒表达载体的抽提
74.从转化的氨苄抗性培养板中挑取单菌落,加至10ml含氨苄抗性的lb培养基中,37℃,220rpm摇床培养18h。取3ml的过夜细菌加至500ml含氨苄抗性的lb培养基中,37℃,220rpm摇床培养12-16h。收获细菌后按照质粒抽提试剂盒(qiagen,货号12262)使用说明书抽提重组慢病毒表达载体,重组慢病毒表达载体1(hpirt-lca),重组慢病毒表达载体2(rft-i-lca)质粒浓度分别达到1000μg/ml。
75.实施例3慢病毒的包装
76.(1)预培养:提前传代293t细胞至150mm培养皿中,置于37℃,5%co2的培养箱中过
夜;第二天观察细胞密度,达到70~80%的汇合率即可进行转染;开始转染之前,使用预热的dmem清洗293t细胞2次,洗涤结束后加入17ml含氯喹的dmem,置于37℃培养箱中备用;
77.(2)转染:准备两个50ml离心管,分别标记为试管1和试管2,向试管1中按顺序加入表2中试剂,轻轻转动混匀;
78.表2试管1中所添加的试剂
79.2m cacl2288μlpspax2(1000μg/ml)11.5μlpmd2g(1000μg/ml)7.5μl重组慢病毒表达载体1或重组慢病毒表达载体2(1000μg/ml)100μlh2o2185μl
80.向试管2中加入2592μl的2x hbs。将试管1中的溶液缓慢滴入试管2,室温静置15min。将混合液滴入步骤(1)含293t细胞的平板中,确保混合均匀,放入37℃,5%co2培养箱中孵育5h,5h后更换为含2%fbs培养基,24h更换新鲜的含2%fbs培养基。
81.(3)收毒:转染后48h收集病毒上清。在48h收毒后补入含2%胎牛血清fbs的新鲜完全培养基,平稳置于37℃,5%co2的恒温培养箱中继续培养,待转染72h后再次收毒。
82.(4)超速离心:将步骤(3)中收集的病毒上清置于50ml离心管,4℃,2000
×
g,离心10min,去除细胞碎片;然后收集病毒原液上清置于超速离心管中,4℃,82700
×
g,离心120min,去掉上清,将慢病毒超速离心后的沉淀重悬于pbs,分装到灭菌处理的病毒管中得病毒原液,-80℃冰箱保存。
83.(5)滴度检测
84.检测前一天,传代293t细胞,每个24孔中加入1
×
105个细胞,体积500ul;
85.按照10倍梯度稀释法稀释步骤(4)所得病毒原液:取6个1.5ml ep管,每管加入完全培养基90ul,然后向第1个ep管中加入病毒原液10ul,充分混匀;吸取第1个ep管中的病毒稀释液10ul,加入第二个ep管中,充分混匀;吸取第二个ep管中的病毒稀释液10ul,加入第三个ep管中,充分混匀,以此类推。将不同病毒稀释样品加入到293t细胞,放入37℃,5%co2培养箱中培养。72h后收取细胞,抽提基因组进行滴度检测。
86.准备转染载体标准品及293t细胞gapdh基因质粒,计算标准品拷贝数:
87.每微升标准品拷贝数=a
×n×
109/m
88.a:标准品质粒的质量(ng)
89.n:阿伏伽德罗常数6.02
×
10
23
90.m:标准品的相对分子质量=标准品的碱基对数
×
1对碱基的平均相对分子质量660
91.例如:109个转染质粒分子,质量约为(109/(6.02
×
10
23
))mol
×
(9000
×
660)g/mol=10ng,
92.109/ml即为10ng/ml;依次稀释直至103/ml;
93.设计ltr及gapdh引物
94.hgapdh-q-f tcaaggctgagaacgggaag(seq id no.4)
95.hgapdh-q-r tcgccccacttgattttgga(seq id no.5)
[0096]5’
ltr-q-f2:ctctggctaactagggaa(seq id no.6)
[0097]5’
ltr-q-r2:cactgactaaaagggtct(seq id no.7)分别以慢病毒感染的细胞基因组和10~107拷贝的标准品为模板,利用上述ltr及gapdh引物进行定量pcr,并根据标准品的ct值和拷贝数绘制标准曲线,将待测样品的ct值代入标准曲线,计算获得待测样品ltr和gapdh的拷贝数。
[0098]
每个细胞整合慢病毒拷贝数=ltr拷贝数
×
2/gapdh拷贝数
[0099]
其中,293t细胞为2倍体,即每个细胞中有2个gapdh内参基因
[0100]
慢病毒(tu/ml)=每个细胞整合慢病毒拷贝数
×
感染细胞数
×
1000/慢病毒体积(μl)
[0101]
检测得到重组慢病毒表达载体1(hpirt-lca)滴度为2
×
107tu/ml;
[0102]
重组慢病毒表达载体2(rft-i-lca)滴度为1
×
107tu/ml。
[0103]
实施例4
[0104]
1、感觉神经元的培养
[0105]
神经细胞选用c57bl/6小鼠的新生幼鼠的背根神经与三叉神经,培养前需将24孔板用多聚赖氨酸与层粘连蛋白平铺。先在每个孔中加入多聚赖氨酸,轻拍培养板使其平铺板底,静置一个小时后将多聚赖氨酸回收,再向每孔中加入层粘连蛋白,轻拍培养板使其平铺板底,放入4℃冰箱备用。
[0106]
将3-5只刚出生c57bl/6小鼠幼崽,解剖取其背根神经节(drg)和三叉神经节(tgn),取出后及时放置于4℃pbs中。25℃,1000rpm,离心5min,弃上清,向tgn、drg中各加入5ml酶溶液(10mg/ml的胶原蛋白酶2.5ml与5mg/ml的分散酶2.5ml),37℃水浴锅中消化30min。当细胞消化至云雾状时,加入5ml dmem终止消化。25℃,1000rpm将细胞离心5min,弃上清,加入完全培养基重悬神经细胞,重悬细胞时应避免产生气泡,可分次重悬,避免对神经细胞的机械损伤。重悬细胞后将细胞悬液过100μm滤膜,使细胞成为单个细胞便于培养。用pbs清洗含层粘连蛋白的24孔培养板,将细胞悬液转至24孔培养板中置于37℃co2培养箱中培养。24h后,更换为含ara-c和ngf的完全培养基,之后每48h给细胞换液,纯化至7天后可进行下一步实验。
[0107]
2、病毒感染神经细胞
[0108]
提前2小时在冰上缓慢解冻慢病毒颗粒,完全解冻后,在使用前用手指轻拨几下,使病毒颗粒分布均匀。
[0109]
用病毒处理神经细胞之前,用预热pbs培养基清洗纯化后的drg或tgn细胞。用不同剂量(5.4
×
104,1.8
×
104,0.6
×
104,0.2
×
104,0.66
×
103,0.22
×
103tu/孔)的病毒感染神经细胞。加入病毒之后细胞置于37℃,5%co2培养箱中培养。24h后向每个孔中加入完全培养基200μl;再过48h后向每个孔中加入完全培养基400μl;再过48h后弃去原始培养基,用预热的pbs清洗细胞,再向每孔加入200μl lk或hk溶液刺激30min使其分泌cgrp,p物质,收获的上清5000rpm,离心5min,取上清存至-80℃备用。再向每孔加入80μl 2
×
lds sample buffer,混合均匀后静置15min,收获底部细胞用作snap-25底物切割检测。其中,lk溶液和hk溶液的组成如下表3所示。
[0110]
表3 lk溶液和hk溶液的组成成分
[0111]
化学药品(相对分子质量)lk溶液hk溶液hepes(238.3)5.36g5.36g
nacl(58.44)7.89g4.59gkcl(74.55)261mg4.47gmgcl2·
6h2o(203.3)203mg203mgcacl2·
2h2o(147)368mg368mgglucose(180.16)595mg595mgbsa(0.1%)1g1gph7.4
ꢀꢀ
加水至1l
ꢀꢀ
[0112]
实施例5
[0113]
一、结果检测
[0114]
采用westernblot的方法分析蛋白样品,具体方法如下:
[0115]
1.将实施例4收获的底部细胞100℃金属浴加热10min,期间涡旋震荡三次冷却至室温备用;
[0116]
2.上样:向电泳槽中倒入电泳缓冲液,取出预制胶(invitrogen,货号nw00120box),撕下底部封条,拔出梳子,用电泳缓冲液清洗上样孔3次,将步骤1准备好的样品按顺序上样;
[0117]
3.电泳:接通电源后,恒压180v电泳;
[0118]
4.转膜:电泳结束后使用转膜装置,20v恒压转膜1h,将蛋白转移到pvdf膜上;
[0119]
5.封闭:用含5%脱脂奶粉的tbst封闭液室温封闭1h;
[0120]
6.一抗孵育:用封闭液稀释snap-25一抗(biolegend,货号836304)与β-tubulin iii(sigma,货号t8578)一抗,4℃孵育过夜;
[0121]
7.洗膜:用tbst清洗pvdf膜3次,每次10min;
[0122]
8.二抗孵育:用tbst稀释对应二抗(jackson,货号115-035-003),室温孵育2h;
[0123]
9.洗膜:用tbst清洗pvdf膜3次,每次10min;
[0124]
10.显影:在暗室里将ecl(thermo,货号32209)滴加到pvdf膜上,反应2-3min后,用g-box曝光拍摄,分析结果。
[0125]
二、结果分析:
[0126]
1.hpirt-lca病毒感染小鼠drg和tgn神经元细胞并切割snap-25
[0127]
用不同剂量(5.4
×
104,1.8
×
104,0.6
×
104,0.2
×
104,0.66
×
103,0.22
×
103tu/孔)的hpirt-lca病毒感染纯化7天的神经细胞(drg和tgn),感染5天后收获样品进行westernblot(其中,选用β-tubulin iii作为内参来检测神经细胞总量的多少),得到附图3。
[0128]
可以看出,正常神经细胞中的snap-25呈现一条大小为25kda的条带,hpirt-lca病毒感染的神经细胞中的snap-25被切割,显示两条相差9个氨基酸的条带。随着病毒剂量的增加,snap-25被切割的量越多,snap-25被切割程度呈现浓度梯度性变化。
[0129]
2.rft-i-lca病毒感染小鼠drg和tgn神经元细胞并切割snap-25
[0130]
用不同剂量(5.4
×
104,1.8
×
104,0.6
×
104,0.2
×
104,0.66
×
103,0.22
×
103tu/孔)的rft-i-lca病毒感染纯化7天的神经细胞(drg和tgn),感染5天后收获样品进行westernblot(其中选用β-tubulin iii作为内参来检测神经细胞总量的多少),得到附图4。
[0131]
可以看出,正常神经细胞中的snap-25呈现一条大小为25kda的条带,rft-i-lca病毒感染的神经细胞中的snap-25被切割,显示两条相差9个氨基酸的条带。随着病毒剂量的增加,snap-25被切割的量越多,snap-25被切割程度呈现浓度梯度性变化。
[0132]
实施例6
[0133]
1.hpirt-lca病毒感染小鼠drg,tgn神经元细胞后p物质的分泌被抑制呈现浓度梯度性变化
[0134]
用不同剂量(5.4
×
104,0.2
×
104tu/孔)的hpirt-lca病毒感染纯化7天的神经细胞(drg和tgn),感染5天后弃去培养基,每孔分别加入200μl lk或hk溶液,37℃培养箱孵育30min,离心后取上清根据elisa试剂盒(cayman,货号583751)说明书的方法步骤检测p物质含量。hk引起神经元去极化分泌的p物质量=(hk上清的p物质量)-(lk上清的p物质量),从而得到附图5。
[0135]
由附图5可以看出,当神经细胞感染最大剂量病毒时(5.4
×
104tu/孔),hk刺激p物质的分泌量相较于正常细胞减少百分之九十左右,病毒感染神经细胞后,抑制了痛觉神经递质的分泌。
[0136]
2.验证hpirt-lca病毒感染小鼠drg神经元细胞具有长效性
[0137]
用不同剂量(5.4
×
104,0.2
×
104tu/孔)的hpirt-lca病毒感染纯化7天的drg神经元细胞,接毒当天记为第0天,为验证hpirt-lca病毒感染神经细胞具有长效性,分别在第7、14、21、28天收获蛋白样品。首先弃去原有培养基,再用pbs轻轻清洗每个孔,再向每孔中加入80μl 2
×
lds sample buffer。按实施例5所述westernblot方法分析snap-25被切割情况,得到附图6。
[0138]
由附图6可以看出,第7、14、21、28天样品中的snap-25蛋白均被切割,表明hpirt-lca病毒持续起作用,目的基因lc/a稳定表达在神经细胞中。
[0139]
实施例7
[0140]
应用实例
[0141]
李某某,女性,65岁,患有右腿膝骨关节炎,行走时有关节疼痛不适感。向患者右腿膝关节内注射50μl,1x10
10 tu/ml的hpirt-lca慢病毒,结果显示:注射7天后,患者关节疼痛感有明显的改善;注射3个月后,患者行走时的关节疼痛感基本消失。
[0142]
张某某,男性,70岁,患有带状疱疹后遗神经痛,卡马西平和苯妥英钠等药物对该患者神经痛没有明显效果。向该患者皮下多部位注射共100μl,1x10
10 tu/ml的hpirt-lca慢病毒,根据疼痛视觉模拟评分法和利兹神经病理性症状和体征进行评分,结果显示:注射5天后,患者神经痛明显改善;注射1个月后,患者神经痛感基本消失,生活质量明显提高。
[0143]
刘某某,男性,69岁,右侧颈部患有鳞状细胞癌,经癌组织切除和放射化疗后,患有严重的局灶性疼痛(视觉模拟评分》6),向患者疼痛区域皮下注射80μl,1x10
10 tu/ml的hpirt-lca慢病毒,根据疼痛视觉模拟评分法进行评分,结果显示:注射3天后,患者疼痛明显改善;注射1个月后,患者痛感基本消失,生活质量明显提高。
[0144]
由以上实施例可知,本发明合成了人源pirt基因启动子序列和兔源β-半乳糖苷α1,2-岩藻糖基转移酶基因(rft-i)最小启动子序列,并将切割snare的蛋白酶编码序列分别插入到这两个启动子下游,构建了两种能特异抑制初级感觉神经元中snare功能的慢病毒载体,分别与pspax2、pmd2.g两种载体共转染293t细胞,制备两种不同的重组慢病毒。慢病
毒感染培养的背根神经节和三叉神经节神经元,有效切割了snare蛋白,抑制了痛觉神经递质(p物质)的分泌。本发明的慢病毒载体具备特异性抑制外周痛觉发生和信号传导的特点,实现基因治疗慢性疼痛的目的。
[0145]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1