一种由3-蒈烯制备1,8-桉叶素衍生物的方法与流程
一种由3
‑
蒈烯制备1,8
‑
桉叶素衍生物的方法
技术领域
1.本发明涉及一种由3
‑
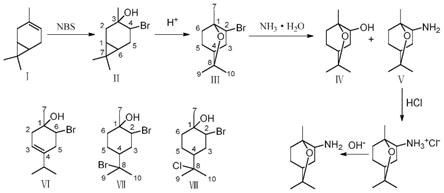
蒈烯制备1,8
‑
桉叶素衍生物的方法,尤其是一种可以同时制备1,8
‑
桉叶素分别含卤素、羟基和氨基三种不同基团的衍生物的方法。
背景技术:
2.1,8
‑
桉叶素是一种重要的含氧单萜化合物,具有抗菌、消炎、杀虫、抗氧化等突出活性,广泛应用于食品、日化、医药等行业中(中国现代医药杂质,2014, 16(4):97
‑
100)。桉叶素的衍生物也被证明具有广泛的生物活性,barton等首先合成3
‑
羟基
‑
1,8
‑
桉叶素,在此基础上又制备得到一系列1,8
‑
桉叶素的酯类衍生物,并测定了它们的除草活性,结果表明这些化合物对于黑麦草和小萝卜都是效果良好的芽后除草剂(journal of pest science,2014,87(3):531
‑
541;journal ofagricultural and food chemistry,2010,58,10147
–
10155)。villecco等通过在1,8
‑ꢀ
桉叶素的3、5、9位引入基团,合成了一系列醇、酯、酮、内酯化合物,并测定了合成化合物的抗菌活性,结果表明两种1,8
‑
桉叶素内酯衍生物对格兰氏阴性菌表现出较高的抑制活性(natural product communications,2008,3(3):303
‑
312)。 2018年巴斯夫公司向欧盟和澳大利亚同时申请登记了两个全新的除草剂,其中一个为cinmethylin,商品名luximo,中文名称为环庚草醚。cinmethylin作用机理独特,通过抑制脂肪酸硫酯酶而发挥药效(pesticide biochemistry andphysiology,2018,148,116
‑
125)。据巴斯夫公司信息,cinmethylin预计将于2020 年在澳大利亚、2021年在英国上市。如下式所示,cinmethylin是1,4
‑
桉叶素的醚类衍生物,由此可以合理推断桉叶素的醚类衍生物具有重要的用途,而醇类或卤素类衍生物是制备醚类衍生物的关键原料。
[0003][0004]
目前,桉叶素醇类衍生物的合成主要有两种途径,一种为化学法,另一种为生物法。化学法是桉叶素在乙酸氧铬(the journal of organic chemistry,1987,52, 1505
‑
1511)、间氯过氧苯甲酸(phytochemistry,1988,27(12):3861
‑
3869)、三氧化铬(australian journal of chemistry,2005,58,785
–
791)等氧化剂作用下,先生成酮类衍生物,再还原得到醇类衍生物,此方法的主要问题是反应会在2、3、5、 6、9、10等位置同时发生,产物往往为多种醇的混合物,单一产物的选择性通常不超过25%,而且混合物难以实现有效分离。生物法是通常各种酶的作用,使 1,8
‑
桉叶素转化为目标产物,目前证明有效的酶有:gymnopilus spectabilis 7423 (biocatalysis,2015,1,44
‑
48)、mucor ramannianus和aspergillus niger(brazilianjournal of microbiology,2015,46(1):
261
‑
264)、细菌p450变体(archives ofbiochemistry and biophysics,2019,663(15):54
‑
63),主要产物皆为3
‑
羟基
‑
1,8
‑
桉叶素和2
‑
羟基
‑
1,8
‑
桉叶素;pleurotus ostreatus和favolus tenuiculus(foliamicrobiologica,2016,61(2):149
‑
157)、rhodococcussp.(electronic journal ofbiotechnology,2006,9(3):232
‑
236),主要产物为2
‑
羟基
‑
1,8
‑
桉叶素和2
‑
羰基
‑
1,8
‑ꢀ
桉叶素。从文献发表年代可以看出,新的研究方向以采用生物法为主,原因是生物法可以保证较高的转化率和选择性,而且大多数时候产物的立体选择性都高于化学法,但是生物法的缺点就是需要对酶进行大量的筛选工作,而且反应过程用时都很长,至少需要培养几天的时间。
[0005]
从目前文献报道来看,1,8
‑
桉叶素的卤素取代衍生物的合成研究相对较少, carman等在
‑
50℃下,采用光照的方式使1,8
‑
桉叶素与氯气发生反应,产物为2、 3、4、7、9位氯取代1,8
‑
桉叶素的混合物(australian journal of chemistry,1983, 36,1483
‑
1493)。bettadaiah等在研究由β
‑
溴
‑
醇类化合物制备烯丙基溴化合物的过程中,发现2
‑
溴
‑
1,8
‑
二羟基
‑
对孟烷在bf3·
et2o作用下可以生成2
‑
溴
‑
1,8
‑
桉叶素,反应需要在苯溶液中回流2h,得率为50%(synthetic communications,2003,33(20): 3615
‑
3620)。相较于羟基和卤素,关于1,8
‑
桉叶素的胺类衍生物报道更加少见。
[0006]3‑
蒈烯广泛存在于松树、白胡椒、九里香、佛手、圆叶当归等植物精油中,其中松节油是其主要来源。3
‑
蒈烯在松节油中的含量与松种有很大关系,印度长叶松(pinus longifolia)和欧洲赤松(pinus sylvestris linn.)所产松节油中3
‑
蒈烯含量可达到50%以上,其余有樟子松((pinus sylvestris var.mongol)、高山松(pinusdensat)、萌芽松(pinus echinat)等,在这些松种所产松节油中含量都超过10%,所以就产量而言,3
‑
蒈烯是仅次于蒎烯的松节油源单萜烯,鉴于松节油作为全球产量最高的天然精油,3
‑
蒈烯作为反应原料在价格上比1,8
‑
桉叶素或其它诸如对孟烷醇等单萜衍生物具有更高优势。1,8
‑
桉叶素的醇、酮、酯类衍生物已被证明具有良好的生物活性。另外,大量研究表明单萜烯的卤素和胺类衍生物具有抗菌、杀虫、除草等广谱生物活性(silva lusitana,2011,19,127
‑
133;林产化学与工业, 2019,39(2):1
‑
8)。而且众所周知羟基、卤素和氨基都具有非常高的反应活性,是制备酯、醚、仲胺、酰胺、希夫碱等一系列结构更复杂、功能更多元的化合物不可缺少的原料。以3
‑
蒈烯为原料制备1,8
‑
桉叶素衍生物,既可以提高松节油资源的应用价值,又可以开发多种新的生物活性物,尤其是潜在天然农药,具有良好的研究与应用双重价值。
技术实现要素:
[0007]
本发明以3
‑
蒈烯为原料合成了2
‑
溴
‑
1,8
‑
桉叶素、2
‑
羟基
‑
1,8
‑
桉叶素和2
‑
氨基
ꢀ‑
1,8
‑
桉叶素。考虑到溴、羟基和氨基都具有非常好的反应活性,通过已有成熟工艺很容易制备得到酯、醚、酰胺、希夫碱等化合物,所以本发明实际上提供了一种新的具有广泛适用性的2位取代1,8
‑
桉叶素衍生物的制备方法。
[0008]
本发明具体内容为:一种由3
‑
蒈烯制备1,8
‑
桉叶素衍生物的方法,
[0009]
第一步,制备以3
‑
羟基
‑4‑
溴
‑
蒈烷(化合物ⅱ)为主要组分的产物a:3
‑
蒈烯 (化合物ⅰ)与n
‑
溴代丁二酰亚胺(nbs)反应生成3
‑
羟基
‑4‑
溴
‑
蒈烷(化合物ⅱ);
[0010]
第二步,制备2
‑
溴
‑
1,8
‑
桉叶素(化合物ⅲ):3
‑
羟基
‑4‑
溴
‑
蒈烷在酸性化合物作用
下,生成2
‑
溴
‑
1,8
‑
桉叶素(化合物ⅲ);
[0011]
第三步,2
‑
溴
‑
1,8
‑
桉叶素与氨水反应生成2
‑
羟基
‑
1,8
‑
桉叶素(化合物ⅳ)和 2
‑
氨基
‑
1,8
‑
桉叶素(化合物
ⅴ
);
[0012]
第四步,第三步反应产物中加入有机溶剂进行萃取,往有机相层滴加酸至白色沉淀不再增加,过滤分离液体部分和固体部分;液体部分回收溶剂,得到2
‑ꢀ
羟基
‑
1,8
‑
桉叶素(化合物ⅳ);
[0013]
第五步,将第四步所得固体加水溶解,往水溶液中加入碱至溶液呈碱性,再加入有机溶剂萃取,有机相层回收溶剂,得到2
‑
氨基
‑
1,8
‑
桉叶素(化合物
ⅴ
)。
[0014]
第二步的化合物ⅲ制备工艺为:将产物a与非亲核性溶剂一起加入反应瓶中,边搅拌边加入酸性化合物,室温下反应一定时间,旋蒸回收溶剂,得到以化合物ⅲ为主要组分的产物b,对产物b进行分离,得到gc含量大于90%的化合物ⅲ。。
[0015]
产物a与非亲核性溶剂加入量比为1∶5g/ml,酸性化合物加入量为反应产物a中化合物ⅱ摩尔质量的1.0~3.0倍,反应温度为10℃~30℃,反应时间为30min~20h。
[0016]
所述的非亲核性溶剂为三氯甲烷、二氯甲烷、四氯化碳、甲苯中的任一种。
[0017]
所述的酸性化合物为三甲基溴硅烷(tmsbr)、三甲基氯硅烷(tmscl)、 hbr、hcl其中一种。
[0018]
化合物ⅳ和化合物
ⅴ
制备工艺为:将gc含量大于90%的化合物ⅲ与氨水一起加热反应一定时间,反应在常压下进行或在密封容器中进行,氨水的加入量为化合物ⅲ质量的4~8倍,反应温度为45℃~70℃,反应时间为1h~3h。
[0019]
所述化合物ⅳ和化合物
ⅴ
都是两种立体异构体的混合物。
[0020]
第四步和第五步所用有机溶剂为乙酸乙酯、乙醚中的一种。
[0021]
产物b除主成分化合物ⅲ外,当所用酸性化合物含溴时主要副产物为2
‑
溴
‑1‑
羟基
‑4‑
对孟烯(化合物
ⅵ
)和2,8
‑
二溴
‑1‑
羟基
‑
对孟烷(化合物
ⅶ
);当所用酸性化合物含氯时主要副产物为2
‑
溴
‑1‑
羟基
‑4‑
对孟烯(化合物
ⅵ
)和2
‑
溴
ꢀ‑8‑
氯
‑1‑
羟基
‑
对孟烷(化合物
ⅷ
)。
[0022]
有益效果
[0023]
1.本发明可同时制备得到3种具有高反应活性的1,8
‑
桉叶素衍生物,基于本发明成果,结合成熟的反应工艺,可制备一系列结构更复杂的1,8
‑
桉叶素衍生物。
[0024]
2.本发明原料3
‑
蒈烯天然储量丰富、价格相对低廉;
[0025]
3.本发明反应过程条件温和(多步反应在室温下即可进行)、用时短、所用试剂皆为常规化学产品,不需要进行专业处理。
[0026]
4.本发明具有较高的原料转化率与产物选择性。而且通过酸碱调节即可使产物实现分离,操作简单、收率高。
附图说明
[0027]
图1本发明工艺反应流程图和主副产物结构式。
[0028]
图2 3
‑
羟基
‑4‑
溴
‑
蒈烷(化合物ⅱ)结构鉴定。gc
‑
ms:m/z理论值234.04,实测值234.0;
13
cnmr(100mhz,cdcl3):δ=15.54(c
‑
7)、17.77(c
‑
1)、19.97 (c
‑
6)、21.92(c
‑
9、c
‑
10)、28.56(c
‑
8)、31.97(c
‑
5)、33.09(c
‑
2)、66.05 (c
‑
4)、71.18(c
‑
3)。
[0029]
图3 2
‑
溴
‑
1,8
‑
桉叶素(化合物ⅲ)结构鉴定。gc
‑
ms:m/z理论值232.05,实测值232.1;
13
cnmr(100mhz,cdcl3):δ=22.55(c
‑
5)、23.11(c
‑
7)、25.83 (c
‑
6)、30.40(c
‑
9)、30.43(c
‑
10)、37.99(c
‑
4)、43.89(c
‑
3)、57.80(c
‑
2)、 83.38(c
‑
8)、83.84(c
‑
1)。
[0030]
图4 2
‑
羟基
‑
1,8
‑
桉叶素(化合物ⅳ)结构鉴定。gc:组分1含量79.20%、组分2含量16.49%;gc
‑
ms:m/z理论值170.13,组分1和组分2实测值皆为170.1;
13
cnmr(100mhz,cdcl3):δ=20.40(1
‑
c
‑
9)、20.44(1
‑
c
‑
10)、27.85(1
‑
c
‑
7)、 28.77(1
‑
c
‑
5)、29.49(1
‑
c
‑
3)、34.99(1
‑
c
‑
6)、40.29(1
‑
c
‑
4)、69.76(1
‑
c
‑
2)、 73.04(1
‑
c
‑
8)、73.53(1
‑
c
‑
1);δ=23.23(2
‑
c
‑
9)、23.45(1
‑
c
‑
10)、24.60(2
‑
c
‑
7)、 28.32(2
‑
c
‑
5)、30.74(2
‑
c
‑
3)、36.57(2
‑
c
‑
6)、43.91(2
‑
c
‑
4)、73.53(2
‑
c
‑
2)、 82.51(2
‑
c
‑
8)、82.95(2
‑
c
‑
1)。组分1和组分2对应位置碳的位移几乎呈相似比例移动,符合空间立体异构体特性,具体结构有待进一步分析。
[0031]
图5 2
‑
氨基
‑
1,8
‑
桉叶素(化合物
ⅴ
)结构鉴定。gc:组分1含量58.98%、组分2含量36.15%;gc
‑
ms:m/z理论值169.15,组分1和组分2实测值皆为169.1;
13
cnmr(100mhz,cdcl3):δ=22.94(1
‑
c
‑
9)、23.37(1
‑
c
‑
10)、24.92(1
‑
c
‑
7)、 28.48(1
‑
c
‑
5)、30.43(1
‑
c
‑
3)、36.22(1
‑
c
‑
6)、44.13(1
‑
c
‑
4)、55.51(1
‑
c
‑
2)、81.82(1
‑
c
‑
8)、83.70(1
‑
c
‑
1);δ=20.62(2
‑
c
‑
9)、20.79(1
‑
c
‑
10)、27.83(2
‑
c
‑
7)、 28.76(2
‑
c
‑
5)、30.18(2
‑
c
‑
3)、35.08(2
‑
c
‑
6)、40.90(2
‑
c
‑
4)、49.98(2
‑
c
‑
2)、 72.44(2
‑
c
‑
8)、76.10(2
‑
c
‑
1)。与2
‑
羟基
‑
1,8
‑
桉叶素(化合物ⅳ)一样,化合物
ⅴ
组分1和组分2对应位置碳的位移呈现相同的移动比例。说明羟基和氨基取代溴的反应机理一致,符合本发明工艺反应特性。
[0032]
图6 2
‑
溴
‑1‑
羟基
‑8‑
对孟烯(化合物
ⅵ
)、2,8
‑
二溴
‑1‑
羟基
‑
对孟烷(化合物
ⅶ
) 和2溴
‑8‑
氯
‑1‑
羟基
‑
对孟烷(化合物
ⅷ
)结构鉴定。gc
‑
ms鉴定结果证明三种副产物m/z实测值与理论值一致。
具体实施方式
[0033]
本发明涉及一种由3
‑
蒈烯制备1,8
‑
桉叶素衍生物的方法,尤其是一种可以同时制备1,8
‑
桉叶素分别含卤素、羟基和氨基三种不同基团的衍生物的方法。本发明方法具体操作步骤为:
[0034]
第一步,化合物ⅱ参照cocker报道工艺(tetrahedronletters,1969,51, 4451
‑
4452)制备,具体操作为:将3
‑
蒈烯(化合物ⅰ)、caco3、水、1,4
‑
二氧六环和n
‑
溴代丁二酰亚胺(nbs)依次加入反应瓶中,于室温下搅拌2h。各物料投料比依次为0.1mol∶0.1mol∶50ml∶100ml∶0.15mol;反应液转入水中,过滤除去沉淀,加入足量与水不互溶有机溶剂萃取,弃去水层;有机层加入足量 10%硫代硫酸钠
‑
水溶液洗涤,弃去水层,有机层加入无水硫酸钠干燥;过滤除去干燥剂,旋蒸回收溶剂,得到以3
‑
羟基
‑4‑
溴
‑
蒈烷(化合物ⅱ)为主要组分的产物a。
[0035]
第二步,将产物a与非亲核性有机溶剂一起加入反应瓶中,边搅拌边加入酸性化合物,室温下反应一定时间,旋蒸回收溶剂,得到以化合物ⅲ为主要组分的产物b。产物a与非亲核性溶剂加入量比为1g∶5ml,酸性化合物加入量为反应产物a中化合物ⅱ摩尔质量的1.0~3.0倍,反应温度为10℃~30℃,反应时间为30min~20h。采用柱层析或精馏等常规分离手段对产物b进行分离,得到 gc含量大于90%的2
‑
溴
‑
1,8
‑
桉叶素(化合物ⅲ)。所述的非亲核性有机溶剂为三氯甲烷、二氯甲烷、四氯化碳、甲苯等中的任一种。所述的酸性化合物为
tmsbr、 hcl、hbr、tmscl等其中一种。
[0036]
第三步,将gc含量大于90%的化合物ⅲ与氨水(25%~28%)一起加入反应瓶中,加热反应一定时间。反应可以在常压下进行、也可以在密封容器中进行,氨水的加入量为化合物ⅲ质量的4~8倍,反应温度为45℃~70℃,反应时间为 1h~3h。
[0037]
第四步,加入与水不互溶的有机溶剂对第三步产物进行萃取,弃去水层,往有机相层滴加hcl,有白色沉淀生成,一直滴加至白色沉淀不再增加,过滤分离液体部分和固体部分。液体部分回收溶剂,得到2
‑
羟基
‑
1,8
‑
桉叶素(化合物ⅳ);
[0038]
第五步,将第四步所得固体加水溶解,往水溶液中加入naoh至溶液呈碱性,再加入与水不互溶的有机溶剂萃取,弃去水层,有机相层回收溶剂,得到 2
‑
氨基
‑
1,8
‑
桉叶素(化合物
ⅴ
)。
[0039]
第四步和第五步操作过程所述有机溶剂为乙酸乙酯、乙醚等与水不互溶的常规化学试剂。
[0040]
实施例1制备以3
‑
羟基
‑4‑
溴
‑
蒈烷为主要组分的产物a
[0041]
将10.8g3
‑
蒈烯、8gcaco3、40ml水、80ml1,4
‑
二氧六环和20gnbs依次加入反应瓶中,于室温下搅拌2h;将反应液转入200ml水中,过滤除去沉淀,加入乙酸乙酯(200ml
×
3次)萃取,弃去水层;有机层加入10%硫代硫酸钠
‑
水溶液(200ml
×
3次)洗涤,弃去水层,有机层加入无水硫酸钠干燥;过滤除去干燥剂,旋蒸回收溶剂,得到产物a15.2g,其中化合物ⅱgc含量为71.8%。
[0042]
实施例2制备2
‑
溴
‑
1,8
‑
桉叶素
[0043]
将1g实施例1所得产物a、5ml三氯甲烷一起加入反应瓶中,打开搅拌,加入0.94g三甲基溴硅烷,20℃下搅拌1h;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅶ
gc含量分别为:42.95%、16.18%和13.14%。
[0044]
实施例3制备2
‑
溴
‑
1,8
‑
桉叶素
[0045]
将1g实施例1所得产物a、5ml三氯甲烷一起加入反应瓶中,打开搅拌,加入0.47g三甲基溴硅烷,20℃下搅拌1h;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅶ
gc含量分别为:36.61%、14.68%和6.53%。
[0046]
实施例4制备2
‑
溴
‑
1,8
‑
桉叶素
[0047]
将1g实施例1所得产物a、5ml三氯甲烷一起加入反应瓶中,打开搅拌,加入1.41g三甲基溴硅烷,20℃下搅拌1h;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅶ
gc含量分别为:40.38%、15.88%和9.77%。
[0048]
实施例5制备2
‑
溴
‑
1,8
‑
桉叶素
[0049]
将1g实施例1所得产物a、5ml三氯甲烷一起加入反应瓶中,打开搅拌,加入0.94g三甲基溴硅烷,30℃下搅拌30min;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅶ
gc含量分别为:41.53%、10.37%和7.53%。
[0050]
实施例6制备2
‑
溴
‑
1,8
‑
桉叶素
[0051]
将1g实施例1所得产物a、5ml三氯甲烷一起加入反应瓶中,打开搅拌,加入0.94g三甲基溴硅烷,10℃下搅拌2h;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅶ
gc含量分别为:34.46%、15.49%和5.16%。
[0052]
实施例7制备2
‑
溴
‑
1,8
‑
桉叶素
[0053]
将1g实施例1所得产物a、5ml二氯甲烷一起加入反应瓶中,打开搅拌,加入0.94g三甲基溴硅烷,20℃下搅拌1h;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅶ
gc含量分别为:39.21%、16.06%和9.29%
[0054]
实施例8制备2
‑
溴
‑
1,8
‑
桉叶素
[0055]
将1g实施例1所得产物a、5ml甲苯一起加入反应瓶中,打开搅拌,加入 0.94g三甲基溴硅烷,20℃下搅拌1h;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅶ
的gc含量分别为:37.88%、14.87%和7.98%。
[0056]
实施例9制备2
‑
溴
‑
1,8
‑
桉叶素
[0057]
将1g实施例1所得产物a、5ml三氯甲烷一起加入反应瓶中,打开搅拌,加入0.67g三甲基氯硅烷,20℃下搅拌20h;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅷ
的gc含量分别为:29.94%、16.15%和10.50%。
[0058]
实施例10制备2
‑
溴
‑
1,8
‑
桉叶素
[0059]
将1g实施例1所得产物a、5ml三氯甲烷一起加入反应瓶中,打开搅拌,加入0.34ghcl,20℃下搅拌20h;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅷ
的gc含量分别为:4.85%、7.04%和35.46%。
[0060]
实施例11制备2
‑
溴
‑
1,8
‑
桉叶素
[0061]
将1g实施例1所得产物a、5ml三氯甲烷一起加入反应瓶中,打开搅拌,加入0.50ghbr,20℃下搅拌20h;旋蒸回收溶剂,得到产物b,其中化合物ⅲ、化合物
ⅵ
和化合物
ⅶ
的gc含量分别为:28.44%、11.86%和17.35%。
[0062]
实施例12制备2
‑
羟基
‑
1,8
‑
桉叶素和2
‑
氨基
‑
1,8
‑
桉叶素
[0063]
将1ggc含量为90.17%的化合物ⅲ与4g氨水(25%~28%)一起加入反应釜中,密封后加热到70℃,反应1h;产物中化合物ⅳ和化合物
ⅴ
的gc含量分别为:46.92%和41.69%。
[0064]
实施例13制备2
‑
羟基
‑
1,8
‑
桉叶素和2
‑
氨基
‑
1,8
‑
桉叶素
[0065]
将1ggc含量为90.17%的化合物ⅲ与8g氨水(25%~28%)一起加入反应瓶中,加热到回流(约70℃),反应1h;产物中化合物ⅳ和化合物
ⅴ
的gc含量分别为:53.96%和32%。
[0066]
实施例14制备2
‑
羟基
‑
1,8
‑
桉叶素和2
‑
氨基
‑
1,8
‑
桉叶素
[0067]
将1ggc含量为90.17%的化合物ⅲ与4g氨水(25%~28%)一起加入反应瓶中,加热到回流(约45℃),反应3h;产物中化合物ⅳ和化合物
ⅴ
的gc含量分别为:37.11%和34.37%。
[0068]
实施例15制备2
‑
羟基
‑
1,8
‑
桉叶素和2
‑
氨基
‑
1,8
‑
桉叶素
[0069]
将1ggc含量为90.17%的化合物ⅲ与6g氨水(25%~28%)一起加入反应瓶中,加热到回流(约65℃),反应1h;产物中化合物ⅳ和化合物
ⅴ
的gc含量分别为:58.57%和25.08%。
[0070]
实施例16分离2
‑
羟基
‑
1,8
‑
桉叶素和2
‑
氨基
‑
1,8
‑
桉叶素
[0071]
在实施例11反应产物中加入足量乙酸乙酯,充分振摇,有机相回收大部分溶剂至2ml左右,滴加hcl(36%)有白色沉淀生成,继续滴加至沉淀不增加,过滤分离固体与液体;液体部分再加入少量乙酸乙酯,分去水层,有机层回收溶剂后得到产物0.28g,其中2
‑
羟基
‑
1,8
‑
桉叶素gc含量为85.41%;固体部分溶于水中,加入naoh至溶液呈碱性,加入乙醚萃取,
乙醚相回收溶剂后得到产物 0.21g,其中2
‑
氨基
‑
1,8
‑
桉叶素gc含量为95.13%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1