N-乙酰半胱氨酸衍生物或其可药用的盐、制备方法及用途
本发明涉及生物医药领域,特别涉及n-乙酰半胱氨酸衍生物或其可药用的盐、制备方法及用途。
背景技术:
n-乙酰半胱氨酸(nac)已被证实是一类效果明确的抗氧化剂,在呼吸系统、肝脏、甲状腺等疾病甚至肿瘤治疗中被广泛使用。nac在细胞内脱乙酰基生成cys;后者是体内合成还原型谷胱甘肽(gsh)的关键原料,而gsh是细胞内至关重要的非酶类抗氧化物,同时也是许多抗氧化酶的底物;nac还可以利用结构中的游离巯基直接发挥抗氧化作用;另外有报道称,nac还参与体内蛋白质的s-过硫化,进而调节部分蛋白质的命运及功能,影响氧化应激信号通路。但值得注意的是,游离巯基本身稳定性不理想,易发生二聚化或氧化,这影响了nac给药后的药理作用。为此,可以选用适当的前药策略先将巯基保护,保证其在体外具有理想的稳定性;而在体内又能够释放出活性巯基产物。
技术实现要素:
发明目的:本发明目的是提供n-乙酰半胱氨酸衍生物或其可药用的盐。
本发明另一目的是提供所述n-乙酰半胱氨酸衍生物或其可药用的盐制备方法及医药用途。
技术方案:本发明提供如式(i)所示结构的n-乙酰半胱氨酸衍生物或其可药用的盐,
r1选自:h或c1-c8烷基;
r2选自:h或甲基;
r3选自:h或甲基;
r4选自:h或c1-c4烷基;
ar选自:苯基、x取代苯基、1-萘基或2-萘基;
x选自:氟、氯、溴、碘、氰基、硝基、三氟甲基或甲砜基。
进一步地,r1选自:h、甲基、乙基、正丙基、异丙基、正丁基或异丁基;
r2选自:h或甲基;
r3选自:h或甲基;
r4选自:h、甲基或乙基;
ar选自:苯基或x取代苯基;
x选自:氟、氯、溴、碘、氰基、硝基、三氟甲基或甲砜基。
进一步地,
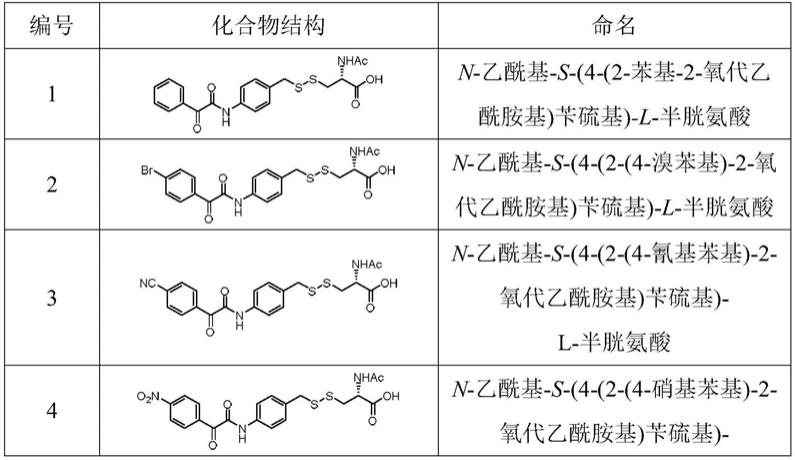
所述如式(i)所示结构的n-乙酰半胱氨酸衍生物或其可药用的盐,为如下任一种:
一种药物组合物,其含有治疗有效量的所述如式(i)所示结构的n-乙酰半胱氨酸衍生物或其可药用的盐,及药学上可接受的载体或辅料。
所述如式(i)所示结构的n-乙酰半胱氨酸衍生物或其可药用的盐的制备方法,合成路线如下所示:
其中,所述r1、r2、r3、r4、ar、x的定义如前;y选自:氯、溴或碘;g选自:
(1)化合物ii发生卤代反应得到化合物iii;
(2)化合物iii经硫脲取代反应得到化合物iv;
(3)化合物iv经脱保护反应得到化合物v;
(4)化合物v与化合物vi偶联,得到化合物i。
进一步地,所述步骤(1)中化合物ii的制备可参照现有文献cn110305036;所采用的溶剂:正己烷、环己烷、苯、甲苯、二氯甲烷、氯仿、四氢呋喃、乙酸乙酯或者用这些溶剂任选组成的混合溶剂;所采用的卤代试剂为氯化氢、氯化亚砜、三氯化磷、溴化氢、三溴化磷或碘化氢;卤代试剂当量为0.1至10当量,优选1至3当量;反应温度为-20℃至60℃,优选温度为0℃至40℃。
进一步地,所述步骤(2)中所采用的溶剂:二氯甲烷、氯仿、乙酸乙酯、乙腈、四氢呋喃、丙酮、2-丁酮、甲醇、乙醇、异丙醇、乙二醇二甲醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、n-甲基吡咯烷酮、水或者用这些溶剂任选组成的混合溶剂;反应温度为-20℃至100℃,优选温度为0℃至50℃。
进一步地,所述步骤(3)中所采用的溶剂:二氯甲烷、乙酸乙酯、氯仿、乙腈、四氢呋喃、丙酮、2-丁酮、乙二醇二甲醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、n-甲基吡咯烷酮、水或者用这些溶剂任选组成的混合溶剂;所采用的脱保护试剂为氨水、乙醇胺、正丁基胺、正己基胺、三乙胺、二异丙基乙基胺、硫代硫酸钠、焦硫酸钠、亚硫酸钠、亚硫酸氢钠、连二亚硫酸钠或用这些试剂任选组成的混合试剂;反应温度为-20℃至100℃,优选温度为20℃至80℃。
进一步地,所述步骤(4)中化合物iv的制备可参照现有文献acsmacrolett.2020,9,606-612及j.am.chem.soc.2020,142,4309-4316;所采用的溶剂:二氯甲烷、氯仿、乙酸乙酯、乙腈、四氢呋喃、丙酮、2-丁酮、甲醇、乙醇、异丙醇、乙二醇二甲醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、n-甲基吡咯烷酮、水或者用这些溶剂任选组成的混合溶剂;所采用的添加剂为:氨水、乙醇胺、正丁基胺、正己基胺、三乙胺、二异丙基乙基胺、乙酸、丙酸、丁酸或苯甲酸;添加剂当量为0至10当量,优选0至3当量;反应温度为-20℃至100℃,优选温度为0℃至50℃。
如式(i)所示结构的n-乙酰半胱氨酸衍生物或其可药用的盐在抗氧化应激的药物中的用途。
有益效果:该类衍生物具有出色的体外稳定性,显著优于nac,且在氧化应激模型中表现出理想药效,优于nac;本发明还提供所述衍生物及其中间体的制备方法、药物组合物及其医药用途。
附图说明
图1为化合物i-4的1hnmr谱图;
图2为化合物i-6的1hnmr谱图。
具体实施方式
实施例1
化合物iii-1的合成:将ii-1(640mg,2.51mmol)置于干燥的圆底烧瓶(50ml),加入二氯甲烷(15ml),在冰浴条件下逐滴滴加pbr3(339mg,1.25mmol)。至原料反应完全,加入饱和碳酸氢钠溶液(30ml),用dcm(3×20ml)萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥。所得粗品经柱层析(石油醚∶乙酸乙酯=2∶1)得到浅黄色固体(702mg,产率88%)并直接用于下一步反应。
化合物v-1的合成:化合物iii-1(636mg,2mmol)、硫脲(183mg,2.4mmol)与thf(20ml)置于干燥的圆底烧瓶(50ml),室温搅拌5h。抽滤得到粗产物iv-1,无需纯化直接进行下一步。将iv-1粗品置于封管中,加入na2s2o5(760mg,4mmol)、二氯甲烷(15ml)与h2o(15ml)。油浴加热至50℃至tlc监测iv-1反应完全。用dcm(3×20ml)萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥。所得粗品经柱层析(石油醚∶乙酸乙酯=2∶1)得到黄色固体v-1(449mg,产率89%)。
化合物i-1的合成:化合物v-1(273mg,1.06mmol)与化合物vi-1(288mg,1.06mmol)置于双颈烧瓶,加入甲醇(10ml)、三乙胺(10mg,10%),室温搅拌过夜;直接减压蒸除溶剂,粗品经柱层析(二氯甲烷∶甲醇=10∶1),得浅黄色固体化合物i-1(292mg,产率64%)。1hnmr(300mhz,dmso)δ12.88(s,1h),10.95(s,1h),8.26(d,j=8.0hz,1h),8.12-7.99(m,2h),7.90-7.52(m,5h),7.35(d,j=8.2hz,2h),4.48(t,j=7.6hz,1h),3.97(s,2h),3.03(dd,j=13.6,4.6hz,1h),2.83(dd,j=13.5,9.0hz,1h),1.87(s,3h).
实施例2
参照实施例1的方法,将实施例1中的起始原料ii-1替换为ii-2,制得化合物i-2。1hnmr(300mhz,dmso-d6)δ12.85(s,1h),10.99(s,1h),8.32(d,j=8.1hz,1h),8.06-7.96(m,2h),7.89-7.79(m,2h),7.76-7.59(m,2h),7.44-7.26(m,2h),4.50(td,j=8.6,4.5hz,1h),3.97(s,2h),3.01(dd,j=13.6,4.7hz,1h),2.82(dd,j=13.6,9.1hz,1h),1.88(s,3h).
实施例3
参照实施例1的方法,将实施例1中的起始原料ii-1替换为ii-3,在步骤(4)中将三乙胺替换为乙酸,将溶剂从甲醇替换为甲醇与四氢呋喃混合溶剂(1∶1v/v),最终制得化合物i-3。1hnmr(300mhz,dmso-d6)δ11.01(s,1h),8.22(dd,j=9.4,2.9hz,3h),8.06(d,j=8.3hz,2h),7.73(d,j=8.4hz,2h),7.35(d,j=8.2hz,2h),4.46(td,j=8.5,4.6hz,1h),3.97(s,2h),3.03(dd,j=13.5,4.7hz,1h),2.84(dd,j=13.5,8.9hz,1h),1.87(s,3h).
实施例4
参照实施例1的方法,将实施例1中的起始原料ii-1替换为ii-4,制备得到化合物iv-4。
化合物v-4的合成:化合物iv-4粗品(445mg,约1mmol)置于圆底烧瓶中,加入正丁胺(176mg,2mmol)与二氯甲烷(15ml)。室温搅拌至tlc监测iv-4反应完全。用dcm(3×20ml)萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥。所得粗品经柱层析(石油醚∶乙酸乙酯=2∶1)得到黄色固体v-4(228mg,产率72%)。
化合物i-1的合成:化合物v-4(220mg,0.7mmol)与化合物vi-1(190mg,0.7mmol)置于双颈烧瓶,加入乙醇(10ml),室温搅拌过夜;直接减压蒸除溶剂,粗品经柱层析(二氯甲烷:甲醇=10∶1),得浅黄色固体化合物i-1(194mg,产率58%)。1hnmr(300mhz,dmso-d6)δ12.89(s,1h),11.03(s,1h),8.45-8.24(m,5h),7.79-7.71(m,2h),7.42-7.31(m,2h),4.51(t,j=8.7,4.6hz,1h),3.99(s,2h),3.03(dd,j=13.5,4.7hz,1h),2.84(dd,j=13.5,9.3hz,1h),1.88(s,3h).
实施例5
参照实施例4的方法,将实施例4中的起始原料ii-4替换为ii-5,制备得到化合物1-5。1hnmr(300mhz,chloroform-d)δ8.31(d,j=8.8hz,2h),8.06(d,j=8.8hz,2h),7.30-7.20(m,2h),7.16-7.03(m,2h),6.51(d,j=7.3hz,1h),4.74-4.65(m,1h),3.81(s,2h),3.51(s,3h),2.76(dd,j=14.1,5.0hz,1h),2.65(dd,j=14.1,6.0hz,1h),2.06(s,3h).
实施例6
化合物i-6的合成:化合物v-4(220mg,0.7mmol)与化合物vi-2(388mg,1.05mmol)置于双颈烧瓶,加入二异丙基乙基胺(13mg,10%)及乙醇(10ml),室温搅拌过夜;直接减压蒸除溶剂,粗品经柱层析(二氯甲烷∶甲醇=10∶1),得浅黄色固体化合物i-6(181mg,产率49%)。1hnmr(300mhz,dmso-d6)δ11.05(s,1h),8.44-8.26(m,5h),7.79-7.68(m,2h),7.34(d,j=8.1hz,2h),4.63(dd,j=8.7,3.0hz,1h),3.96(s,2h),3.65(s,3h),1.92(s,3h),1.35(s,3h),1.33(s,3h).
以上各实施例中的溶剂、还原剂、酰化剂、烷基化试剂等可根据需要替换。
实施例7
体外稳定性的测定
实验方法:
分别取人工胃液、肠液、血浆、pbs(980μl),加入10μl的化合物储备液(100mm),空白对照为pbs。震荡混匀后置于37℃条件下温孵(n=3)。在不同时间点下(t=0、5、15、30、60min)取样100μl,加入500μl乙腈(含内标华法林2ng/ml),震荡30s,4℃离心18000rpm×10min,取上清液200μl,二次离心(18000rpm×10min)后弃沉淀,取上清80μl进样。
色谱条件:
色谱柱:agilentzorbaxsb-c18column(5μm,4.6×250mm);柱温:40℃;水相(a):超纯水(0.1%甲酸),有机相(b):甲醇;流速:0.8ml/min;
梯度洗脱程序如下表:
质谱条件:
实验结果:
实验结果:与nac相比,本发明所公开衍生物均有较好的体外稳定性。
实施例8
体外抗氧化应激活性测定
实验方法:hepg2细胞分组:空白对照组、模型组、给药组。其中空白对照组用正常的细胞培养液培养,模型组用400μm的h2o2培养12h,给药组分别加入化合物预处理24h后,再加入400μm的h2o2培养12h。
细胞培养:取对数生长期hepg2细胞以8×104/ml接种于96孔培养板,每孔100μl。培养基中加入10μlcck-8原液,继续在培养箱孵育,指定时间利用酶标仪在450nm处测定光密度值;计算出其细胞存活率。
实验结果如下表:(n=6)
实验结果:化合物表现出不同程度的抗氧化应激活性,且活性优于nac;尤其是化合物i-4与i-6,其抗氧化应激活性十分显著。
- 还没有人留言评论。精彩留言会获得点赞!