一种岷江百合诱导型启动子PG1及其应用
1
‑
9)。从白粉病病菌(oidium heveae)基因组中分离得到一个wy7启动子,将wy7启动子连接报告基因gus转入烟草中瞬时表达并给与适当的逆境处理,低温和盐胁迫下诱导了wy7驱动的gus基因表达,表明wy7启动子受低温和盐胁迫因子的诱导(wang y, wang c, rajaofera n, et al. wy7 is a newly identified promoter from the rubber powdery mildew pathogen that regulates exogenous gene expression in both monocots and dicots. plos one, 2020, 15(6):e0233911)。
技术实现要素:
5.本发明目的是提供一种诱导型启动子pg1,来源于岷江百合,其核苷酸序列如seq id no:1所示。
6.本发明另一目的是将该启动子应用在基因工程中,即在生物和非生物胁迫下作为诱导表达启动子调控外源基因在转基因受体植物中的特异高效表达。
7.本发明涉及分离诱导型启动子片段并鉴定其表达活性,本发明从岷江百合中克隆获得诱导型启动子,该启动子长904bp。将本发明分离克隆的诱导型启动子片段置换pbi121载体上的camv 35s启动子,由诱导型启动子驱动报告基因gus的表达框,通过根癌农杆菌(agrobacterium tumefaciens)介导将其转入模式植物烟草中表达,并通过进一步实验揭示诱导型启动子的表达特性,为后期利用该启动子调控外源基因在转基因植株中的高效特异表达奠定基础。发明人将这个启动子命名为pg1。
8.将本发明中pg1启动子驱动gus的表达框转入烟草中,采用几种植物激素、生物和非生物胁迫处理转基因烟草植株,并进行gus活性的荧光定量分析,检测结果表明,pg1启动子响应采用几种植物激素、生物和非生物胁迫处理,脱落酸、水杨酸、尖孢镰刀菌(fusarium oxysporum)、致密链格孢(alternaria compact)、稻黑孢霉(nigrospora oryzae)、伤胁迫能明显诱导启动子pg1的活性。
9.上述启动子pg1可以在基因工程中应用于外源基因的诱导表达,具体操作如下:(1)采用扩增pg1的特异引物,从岷江百合幼嫩组织中提取基因组dna,通过聚合酶链式反应(polymerase chain reaction,pcr)扩增出pg1,然后将其连接到pgem
‑
t载体上,经测序获得具有序列正确的克隆;(2)用限制性内切酶酶切pgem
‑
t
‑
pg1载体,回收启动子片段;同时采用合适的限制性内切酶酶切去除植物表达载体上的组成型表达启动子,通过胶回收得到载体大片段;再将所获得pg1片段与pbi121
‑
gus载体片段连接,构建植物诱导表达载体;之后将所构建的植物诱导表达载体通过根癌农杆菌介导转入受体植物中。转基因植株在遭受尖孢镰刀菌、致密链格孢、稻黑孢霉、伤害胁迫时,启动子pg1驱动的目的基因会诱导并上调表达水平,此外,体内体外的脱落酸、水杨酸也会诱导目的基因高水平表达。
10.本发明为植物基因工程应用中提供了一个新的诱导表达的启动子。基因工程中植物超表达载体常用来自花椰菜花叶病毒的35s启动子,该启动子为组成型表达启动子,目的基因的表达大体恒定在一定水平上,在不同组织、部位表达水平没有明显差异,所以转入植物的外源基因的表达不受控制,导致蛋白大量积累且浪费能量。而诱导型启动子可以在植物受到外界胁迫或化学因素影响时提高基因的表达量,在去除胁迫或化学处理后即下调目的基因的表达,可保证在植物受到逆境胁迫时起到保护植物、抵抗外界刺激的效果,反之在
适宜的环境中不浪费植物的能量。此外,在基因工程应用中诱导型启动子不但可以避免目的基因的持续表达对植物能量的过度消耗,而且可以消除基因产物积累对植物本身造成的伤害。几种激素(脱落酸、水杨酸)、非生物胁迫(伤害)、生物胁迫(尖孢镰刀菌、致密链格孢、稻黑孢霉)明显诱导本发明中pg1启动子的表达活性,因此本发明在抗生物或非生物胁迫的基因工程中具有广阔的应用前景。
附图说明
11.图1是本发明中启动子pg1 (a图)和pbi121载体(b图)的胶回收产物检测结果;图2是本发明中pbi121
‑
pg1
‑
gus转化大肠杆菌的阳性克隆检测结果,其中阳性对照是以pgem
‑
t
‑
pg1质粒为模板的pcr反应,阴性对照是以无菌水为模板的pcr反应;图3是本发明中部分pbi121
‑
pg1
‑
gus转基因烟草的pcr筛选结果,其中阳性对照是以质粒pbi121
‑
pg1
‑
gus为模板的pcr反应;wt:非转基因烟草(野生型)总dna为模板进行的pcr反应。
12.图4是本发明中gus酶活测定的标准曲线;图5是本发明中pbi121
‑
pg1
‑
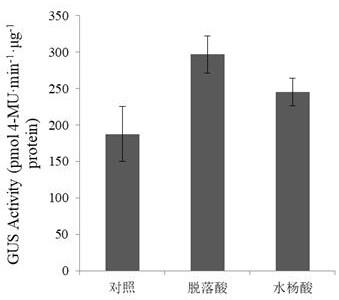
gus转基因烟草在脱落酸、水杨酸处理后的gus活性,其中对照为正常生长的pbi121
‑
pg1
‑
gus转基因烟草的gus活性;图6是本发明中pbi121
‑
pbi121
‑
pg1
‑
gus转基因烟草在伤害处理后的gus活性,其中对照为正常生长的pbi121
‑
pg1
‑
gus转基因烟草的gus活性;图7是本发明中pbi121
‑
pg1
‑
gus转基因烟草在尖孢镰刀菌、致密链格孢、稻黑孢霉接种后的gus活性,其中对照为正常生长的pbi121
‑
pg1
‑
gus转基因烟草的gus活性。
具体实施方式
13.下面通过附图和实施例对本发明进一步说明,但本发明保护范围不局限于所述内容,本实施例中方法如无特殊说明的均按常规方法操作,所用试剂如无特殊说明的采用常规试剂或按常规方法配置的试剂。
14.实施例1:岷江百合诱导型启动子pg1的克隆以及序列分析以提取的岷江百合根基因组dna为模板,用扩增启动子pg1的特异引物(上游引物为:5’gccccatagaccctatccaagta3
’’
,下游引物为:5’caggggcagagggttgac3’,通过pcr克隆启动子pg1的序列。反应体系(20 μl)为岷江百合基因组dna 0.5μg、2μl 10
×
advantage 2 pcr buffer、1.8μl dntp mix (10mm each)、0.2μl 上游引物(10 μm)、0.2μl 下游引物(10 μm)、0.2μl advantage 2 pcr polymerase mix、14.6μl pcr
‑
grade 水。pcr反应条件:94℃ 5min;94℃ 30s,63℃ 30s,72℃ 50s,32个循环;72℃ 5min。pcr结束后,取8μl进行琼脂糖凝胶电泳,用以检测扩增产物的特异性以及大小。
15.所得到pcr产物只有一条dna带,直接对pcr产物进行ta克隆,使用的试剂盒为pgem
‑
t vector system (promega),反应体系和操作过程为:取1.5μl pcr产物,依次加入1μl pgem
‑
t vector (50 ng/μl)和2.5μl 2
×
ligation solution i,混匀后置于16℃过夜反应。通过热激转化法将连接产物转入大肠杆菌dh5α感受态中。用含有氨苄青霉素(ampicillin,amp)的lb固体培养基筛选阳性克隆。挑选若干个单菌落,摇菌后用扩增pg1的特异引物检测多克隆位点插入pg1的克隆。将得到的阳性克隆进行测序,最终获得的启动子
pg1长904bp。采用plantcare对启动子的顺式作用元件进行预测,预测启动子序列中与逆境胁迫相关的顺式作用元件。
16.实施例2:pg1
‑
gus表达载体构建pbi121多克隆位点有hindⅲ和bamhⅰ酶切位点,因此在扩增启动子的特异引物分别添加hindⅲ和bamhⅰ的识别位点。采用sanprep柱式质粒dna小量抽提试剂盒(上海生工)提取插入pg1的大肠杆菌质粒pgem
‑
t
‑
pg1以及植物表达载体pbi121质粒,取1μl用于琼脂糖凝胶电泳以检测所提取质粒的完整性及浓度高低。用限制性内切酶hindⅲ和bamhⅰ分别对质粒pgem
‑
t
‑
pg1和pbi121进行双酶切(5μl体系),反应体系和操作过程为:分别取20μl pgem
‑
t
‑
pg1和pbi121质粒、依次加入10μl 10
×
h buffer、5μl bamhi、5μl hindiii、60μl ddh2o,混匀后短时离心,置于37℃过夜反应。将所有酶切产物进行琼脂糖凝胶电泳,然后使用sanprep柱式dna胶回收试剂盒(上海生工)对启动子片段和pbi121载体大片段分别进行胶回收,取1μl回收产物通过琼脂糖凝胶电泳检测回收片段的大小以及浓度,结果如图1所示。
17.利用t4 dna ligase (takara),将回收的启动子dna片段和pbi121载体片段连接起来,反应体系(20μl),操作过程为:取10μl pg1 dna片段依次加入2μl pbi121载体dna、2μl 10
×
t4 dna ligase buffer、1μl t4 dna ligase、5μl ddh2o,混匀后短时离心,然后16℃水浴过夜反应。接着采用热激转化法将连接产物转入大肠杆菌dh5α中,用含有50mg/l卡那霉素的固体培养基筛选阳性克隆。挑选单菌落摇菌,以菌液为模板用扩增启动子pg1的特异引物进行pcr,挑选出pg1与pbi121成功连接的克隆,在得到的阳性菌株中加入甘油并置于
‑
80℃保存备用。
18.采用sanprep柱式质粒抽提试剂盒提取并纯化上述大肠杆菌dh5α中的pbi121
‑
pg1
‑
gus质粒。随后用液氮冻融法将上述构建的植物表达载体pbi121
‑
pg1
‑
gus转入所制备的根癌农杆菌lba4404感受态细胞中。操作步骤为:取0.2μg pbi121
‑
pg1
‑
gus质粒加入含有200μl感受态细胞的离心管中,轻轻混匀后冰浴5min,随后转入液氮中冷冻1min,然后迅速置于37℃水浴5min,再冰浴2min,之后加入500μl lb液体培养基于28℃振荡培养4h。将活化后的农杆菌涂于含有50mg/l卡那霉素的lb固体培养基上,28℃倒置培养。挑选单菌落摇菌,再用扩增pg1的特异性引物进行pcr反应,检测pbi121
‑
pg1
‑
gus是否转入农杆菌中。对于图2中所示的阳性克隆,加入甘油后置于
‑
80℃保存备用。
19.实施例3:农杆菌介导的植物遗传转化以及转基因植物筛选本实验的转基因受体是烟草,将烟草种子用75%的酒精浸泡30s,无菌水洗涤后用0.1%的hgcl2浸泡8min,然后再用无菌水洗涤若干次,播种于1/2 ms培养基上,28℃暗培养5
‑
8d,发芽后转至光照培养箱(25℃,16h/d光照),以后每月用ms培养基继代一次。
20.将
‑
80℃冰箱中保存的含有pbi121
‑
pg1
‑
gus质粒的农杆菌lba4404菌液取出,取10μl菌液接种于1ml含有20mg/l利福平和50mg/l卡那霉素的lb液体培养基中,28℃、200rpm振荡培养至浑浊。吸取500μl菌液均匀涂布于含有20mg/l利福平和50mg/l卡那霉素的lb固体培养基上,28℃倒置培养至长出菌苔。用接种环刮取3
‑
5环菌苔接种于40ml含25mg/ml乙酰丁香酮的mgl培养基中,28℃、220rpm振荡培养直至od
600
约为0.6。将无菌烟草组培苗叶片剪成约1cm2大小的叶盘,浸泡于含有悬浮农杆菌的mgl培养基中,25℃震荡培养15min。用无菌滤纸将叶盘表面的菌液吸干后转入烟草共培养培养基中,22℃暗培养2天。将共培养后的叶
盘转接到烟草筛选培养基上,培养于光照培养箱(25℃,16h/d光照)。培养约3周,将分化出来的烟草幼苗切下并继代于含有50mg/l卡那霉素和300mg/l头孢霉素的生根培养基上进行生根培养。
21.采用ctab法提取转基因烟草植株叶片的基因组dna,取1μl基因组dna进行琼脂糖凝胶电泳检测其完整性和浓度。以转基因植株的基因组dna为模板用扩增启动子pg1的特异引物进行pcr反应。pcr结束后,取8μl产物用于琼脂糖凝胶电泳以检测阳性转基因植株。部分转基因烟草植株的扩增结果如图3所示,岷江百合诱导型启动子pg1转基因烟草共筛选到29株阳性转基因植株。
22.实施例4:转基因烟草的gus荧光定量检测对转基因烟草叶片gus活性的荧光定量分析参考jefferson等(jefferson r. assaying chimeric genes in plants: the gus gene fusion system. plant mol biol rep. 1987, 5(4): 387
–
405)的方法,其反应机理是:gus可与底物4
‑
mug反应,催化产生4
‑
mu,4
‑
mu在激发波长为365 nm、发射波长为455nm条件下产生荧光,产生的荧光值可以通过荧光分光光度计进行定量测定。
23.将预先处理好的烟草叶片置于装有液氮的研钵中研磨成粉末,加入400 μl gus提取缓冲液,将匀浆转移至1.5ml离心管中,于4℃、12000 g离心10min。离心结束后,收集上清于新的离心管中。预先取1ml的4
‑
mug溶液(1 mmol/l)于2.0ml离心管中37℃预热10min。取50μl上清液加入至预热的gus反应缓冲液中,迅速摇匀,并立即取200μl反应混合液置于1.8ml的终止缓冲液中(操作时间少于30s),作为酶促反应0点样(荧光测定时以此为空白对照),剩余液体继续37℃反应并开始计时。在反应15min、30min、45min时分别取200μl反应混合液,加入至1.8ml终止缓冲液中,用于荧光测定。使用荧光分光光度计在激发波长为365nm,发射波长为455 nm的条件下测定各样品的荧光值。制作4
‑
mu标准曲线:将1mm 4
‑
mu母液用反应终止液分别稀释成5nm、10nm 、20nm、40nm、60nm,80nm和100nm的不同梯度液,在激发波长为365nm,发射波长为455nm的条件下测定各梯度液的荧光值,以反应终止液为空白对照,用测得的荧光值和4
‑
mu的浓度绘制标准曲线(如图4所示)。取10μl上清液,采用改良的考马斯亮蓝法测定样品的蛋白含量。以一分钟催化4
‑
mug生成1pmol 4
‑
mu的酶量为一个活力单位,gus酶活以每μg总蛋白的酶活力计算,即表示为4
‑
mu pmol/min/μg(蛋白)。通过标准曲线,计算出转基因烟草的gus活性。
24.为了检测岷江百合启动子pg1对植物激素、生物胁迫与非生物胁迫的响应,分别用几种植物激素、生物胁迫和非生物胁迫因子处理转基因烟草的叶片,并通过上述方法测定处理前后的gus活性,以正常生长未处理的转基因烟草叶片gus活性作为对照。如图5,在脱落酸、水杨酸处理后,岷江百合启动子pg1转基因烟草叶片的gus活性明显上调,对启动子活性的诱导程度来看,脱落酸>水杨酸。受害处理后转基因烟草的gus活性如图6所示,伤害这种非生物胁迫因子显著上调启动子pg1的活性。用三种病原真菌尖孢镰刀菌、致密链格孢、稻黑孢霉接种转基因烟草的叶片,均显著上调启动子pg1的活性(图7),从诱导程度上来看,致密链格孢>稻黑孢霉>尖孢镰刀菌。上述实验结果表明,岷江百合启动子pg1响应几种植物激素、非生物胁迫及生物胁迫的处理,脱落酸、水杨酸、尖孢镰刀菌、致密链格孢、稻黑孢霉、伤胁迫能明显上调pg1驱动的gus活性。显然,岷江百合启动子pg1是一种植物激素、生物胁迫和非生物胁迫因子诱导型启动子,可应用于植物抗逆基因工程。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1