本发明属于有机合成领域,具体涉及一种3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的合成方法。
背景技术:
3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮是合成盐酸贝那普利的重要中间体。盐酸贝那普利是瑞士giba-geigy公司开发的一种血管紧张素转换酶抑制剂(acei),主要用于治疗高血压、充血性心力衰竭,具有不良反应少,安全性好的优点。盐酸贝那普利为国家医保乙类产品,是目前中国市场上使用最为广泛的血管紧张素转换酶抑制剂(acei),为我国临床上治疗高血压的一线药物。
合成3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的关键中间体为为α-四氢萘酮。文献报道的合成α-四氢萘酮的主要方法有三种:
1,以苯丁酸或者苯丁酰氯为起始原料,经过环化反应生成α-四氢萘酮该策略起始原料价格较贵,限制了其应用。
2,以四氢萘为起始原料,以氧气和/或空气为氧化剂,催化体系为三价铁离子,硝酸根离子和n-羟基邻苯二甲酰亚胺三者联用(cn111606791)或者n-羟基邻苯二甲酰亚胺及亚硝酸酯联合催化(cn107011133),但该合成策略一般会有羟基及多氧化产物生成,需要柱层析纯化,这也限制了其规模化生产。
3,以γ-丁内酯及苯为起始原料,在alcl3催化下合成α-四氢萘酮。该策略原料价格便宜易得,工艺也适合工业化生产。
以α-四氢萘酮为起始原料,合成3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的合成策略大致分为两种:一种是先成肟,然后进行贝克曼重排反应,最后进行溴化反应得到目标产物。另一种是先进行溴化、成肟反应,随后进行贝克曼重排反应得到目标产物。
1、以α-四氢萘酮为起始原料,先进行成肟反应,随后进行贝克曼重排,最后进行溴化反应得到目标产物。如文献bio.med.chem.2015,25,3488,us6777550,us4575503,us4410520及us4473575所述。
路线1
2、以α-四氢萘酮为起始原料,先进行溴化、成肟反应,最后进行贝克曼重排得到目标产物。如文献journalofheterocyclicchemistry,1994,31,1545,cn105017154,us5677297所述。
路线2
以上两种策略在溴化时,都会用到大量的液溴,首先液溴在使用过程中易挥发,有强烈的毒害性和腐蚀性,会刺激眼睛和呼吸道黏膜。液溴的使用过程中,对设备和人员也提出了更高的要求。其次,液溴反应结束会产生一分子氢溴酸,使废液呈强酸性,这也给废液处理增加了难度。
因此,本领域需要开发出新的,更温和高效的制备3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的方法。
技术实现要素:
本发明所要解决的技术问题是,3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮现有合成方法中存在的后处理麻烦,收率较低,环境污染大,成本较高等其中一项或多项技术问题。
本发明的技术方案是,提供3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的合成方法。
本发明提供一种3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的合成方法,以化合物α-四氢萘酮为前体,分别经过溴化、成肟及贝克曼重排反应得到3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮。其具体合成路线为:
具体包括以下步骤:
1)α-四氢萘酮的合成
向苯中加入三氯化铝,搅拌下加入γ-丁内酯,加毕回流至反应完全,反应完毕后,加入盐酸溶液水解,搅拌0.5-3小时后分层,有机层分别水洗、碱洗、水洗至中性,旋蒸除去苯即可得到粗品,粗品减压蒸馏即可得到α-四氢萘酮;
2)2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺的合成
s1.反应瓶中依次加入α-四氢萘酮,hbr溶液、h2o及甲醇,然后向其中加入oxone,30-35℃反应至完全,静置分出下层有机相;s2.向有机相加入甲醇,然后加入硫酸羟胺,投毕,加水稀释,然后25-40℃保温反应60-90小时,保温反应结束后投入小苏打,25-40℃保温反应40~60小时,过滤,滤饼用水洗,再用甲醇淋洗,得到2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺粗品备用;
3)3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的合成
往反应瓶中加入多聚磷酸,加热升温至60-80℃备用,另取反应瓶加入2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺粗品和甲苯加热到60-85℃溶解,保温静置0.5-1小时后分出水相,甲苯相直接加入上述多聚磷酸液体中,保持75℃-90℃搅拌反应,反应完毕后,冷却至室温后倒出甲苯相,加水,温度保持在20-40℃,搅拌1~3小时,过滤,滤饼分别水洗、饱和碳酸氢钠洗、水洗,滤饼抽干得3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮粗品;
4)3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的精制
粗品用甲醇溶解后加入活性炭脱色,脱色母液过滤,除去甲醇至一半后降温到低于10℃重结晶,过滤后滤饼烘干得到3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮纯品。
优选地,γ-丁内酯,苯,无水三氯化铝的摩尔比为1:(8~10):(2~3)。
优选地,步骤1)中,盐酸溶液的浓度为10wt%,盐酸溶液的加入量为苯的质量的1~2倍。
优选地,步骤2)中,α-四氢萘酮,hbr及oxone的摩尔比为:1:2:(2-2.5),α-四氢萘酮、硫酸羟胺和小苏打的摩尔比为1:1:1.5。
优选地,s1步骤中,水和甲醇的加入质量比为8:9,s2步骤中,水和甲醇的加入质量比为3:5。
优选地,步骤3)中,多聚磷酸的加入量为2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺的3-8倍重量,加入的甲苯和水的体积比为3:4。
优选地,活性炭的加入量为3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮粗品质量的15~20%。
本发明的有益效果主要体现在:操作简单,三废产生少,环境污染小,更适合工业化生产。
本发明的优势体现在2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺的合成过程中。以hbr取代液溴,降低了工艺操作的难度,同时减少了酸性废水的产生,并可以实现溶剂的循环利用。
附图说明
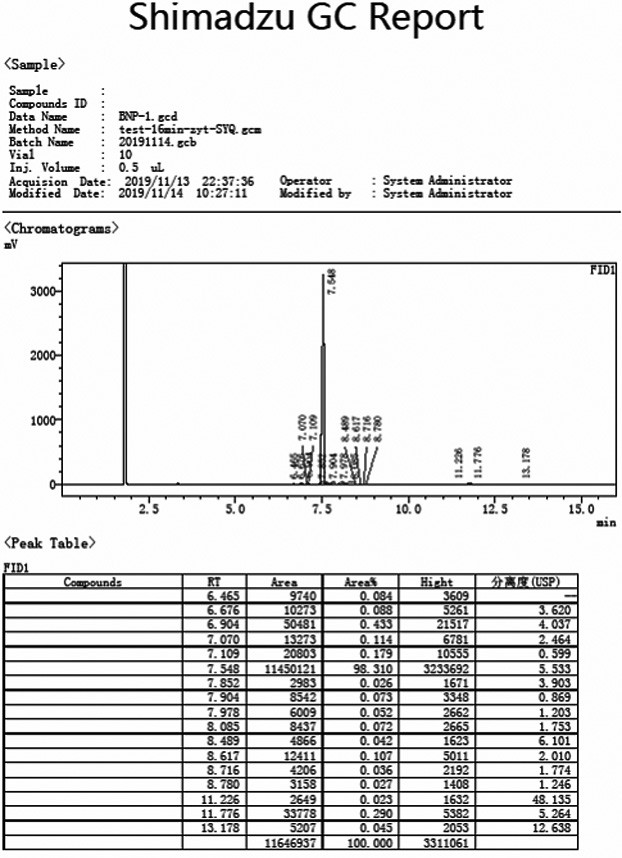
图1是实施例1中α-四氢萘酮的gc谱图;
图2是实施例1中2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺粗品的hplc谱图;
图3是实施例1中3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的hplc谱图;
图4是实施例1中α-四氢萘酮1hnmr谱图;
图5是实施例1中3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的1hnmr谱图。
具体实施方式
为了进一步说明本发明的内容及特点,下面结合实施例进一步说明。
实施例1:
一种3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的合成方法,过程如下:
(1)α-四氢萘酮的合成
在反应瓶中加入苯(1500g,19.2mol)然后分批加入三氯化铝(774g,5.81mol),搅拌下加入γ-丁内酯(200g,2.32mol),加毕80℃回流反应24小时。备好10wt%的盐酸溶液2500g,然后分3批加入反应料液,加毕,搅拌1.5小时后,分层,水层为三氯化铝废水,有机层分别以水洗,饱和碳酸氢钠水溶液洗,最后水洗至中性。旋蒸除去苯即可得到粗品(粗品为液体)345g。粗品减压蒸馏即可得到α-四氢萘酮(260g,1.78mol),其gc图如图1所示,通过图1可知α-四氢萘酮gc纯度为98.3%,α-四氢萘酮1hnmr谱图如图4所示,1hnmr(400mhz,cdcl3):δ8.02(d,1h,j=7.6hz),7.48-7.44(m,1h),7.29(t,1h,j=7.2hz),7.24(d,1h,j=8.0hz),2.96(t,2h,j=6.0hz),2.64(t,2h,j=6.4hz),2.16-2.10(m,2h).
(2)2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺的合成
反应瓶中依次加入α-四氢萘酮260g(1.78mol),hbr(48%,h2o,3.56mol),水(800g)和甲醇(900g),室温下分3批加入oxone(2407g,3.92mol),控制温度33-35℃开始72小时保温反应。反应完毕,静置分出下层有机相。向其中加入甲醇(1500g),加入硫酸羟胺(183g,1.78mol)。投毕,加水900ml稀释,然后控制30-32℃开始85小时保温反应。保温反应结束投入小苏打(224g,2.67mol),并控制30-32℃继续保温反应50小时。冷却至10℃,过滤,滤饼用水洗,再用30%(v/v)甲醇淋洗,得到2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺湿品固体1500g(以α-四氢萘酮1.78mol计,得到约425g2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺)备用,其hplc谱图如图2所示,由图2可知,2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺粗品纯度为87.8%。
(3)3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的合成
往反应瓶中加入多聚磷酸(1700g,4倍重量),加热升温至65℃备用,另取反应瓶加入2-溴-3,4-二氢-n-羟基-(2h)-萘亚胺湿品1500g,甲苯9l加热到80℃溶解,保温静置1小时后分出水相,甲苯相直接加入上述多聚磷酸液体中,保持85℃搅拌反应。反应完毕后,冷却至室温后倒出甲苯相,加水12l,温度保持在35℃左右,搅拌2小时,过滤,滤饼分别用水、饱和碳酸氢钠、水各洗1次,滤饼抽干得3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮湿品280g,烘干得粗品225g.
(4)3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮的纯化
粗品用甲醇(3l)溶解后加入活性炭35g脱色,脱色母液过滤,然后除去甲醇至一半后降温到低于10℃重结晶,过滤后滤饼40℃常压烘干得到3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮纯品(191.5g,0.80mol),hplc谱图如图3所示,由图3可知,产品纯度为99.5%,3-溴-1,3,4,5-四氢-2h-1-苯并氮杂卓-2-酮1hnmr谱图如图5所示,1hnmr(400mhz,dmso):δ10.09(s,1h),7.30(dd,2h,j=9.2hz,7.6hz),7.18(t,1h,j=7.6hz),7.05(d,1h,j=8.0hz),4.62(dd,1h,j=10hz,7.6hz),2.85-2.75(m,3h),2.56-2.52(m,1h).
上述的实施例结果可用来进一步说明本发明,但并不能以此限制本发明的保护范围。凡根据本发明实质所做的修饰和等效变换,均应包含在本发明专利的权利要求范围之内。