一种烟草生殖细胞特异高表达基因及应用的制作方法
1.本发明涉及生物技术领域,具体涉及一种烟草生殖生殖细胞特异高表达基 因及应用。
背景技术:
2.crispr/cas9系统的原理是通过由crrna与tracrrna形成的sgrna (single
‑
guide rna,sgrna)识别靶位点,并引导cas9对靶基因进行定向剪 切,cas9是由hnh和ruvc两个结构域组成,两个结构域分别负责切割dna的 两条链,形成dna双链断裂(double
‑
strand break,dsb)。而dsb主要通过 两种方法进行修复。一种是非同源末端连接(nonhomologous end joining, nhej),这种修复方式会引起碱基的插入和缺失从而导致基因功能缺失。当有 供体dna模板存在时,同源重组(homologous recombination,hr)的精确修 复方式就会发生,但其效率较低。crispr/cas9基因编辑系统设计简单,操作 容易,成本较低,得到了科研人员和实验室的“追捧”,因此,crispr/cas9 系统也得到了很好的推广和发展。在2013年crispr/cas9基因编辑体系率先 成功在水稻、拟南芥、烟草和小麦原生质体和农杆菌介导的cas9
‑
sgrna共骨 架载体稳定转化。随后在马铃、大豆、玉米、大麦等植物中进行了该体系的应 用,并且在拟南芥、水稻、烟草中还实现了多基因的同时编辑。
3.crispr/cas9系统在植物中通常是利用农杆菌介导的遗传转化使其整合 到基因组中发挥作用,构建的植物基因编辑载体通常利用花椰菜花叶病毒的 35s启动子启动表达cas9。已有的研究结果显示,这种常规的基因编辑载体, 获得的后代编辑效率低。2015年,研究人员发现利用拟南芥中的yao基因启 动子驱动cas9,t2代的转基因拟南芥突变效率达到40.9%
‑
85.7%,并且在t2 代中出现了靶标基因发生突变但没有cas9嵌入的植株。在同一年,研究人员 利用拟南芥中的卵母细胞特异性基因ec1.2的启动子驱动cas9,称为epc(theegg cell
‑
specific promoter
‑
controlled crispr/cas9 system)系统,高效 的获得了多个目标基因的非嵌合t1代突变体。
4.作为重要的经济作物和模式植物之一的烟草(nicotiana tabacum),有着 重要的研究价值。但是由于栽培烟草是异源四倍体且基因组庞大复杂,在利用 常规crispr/cas9基因编辑载体进行编辑时,后代植株出现嵌合的比例较高且 基因编辑效率低,在应用过程中耗费大量的人力、物力、财力。
技术实现要素:
5.本发明的目的是提供一种烟草生殖细胞特异表达基因及应用,以解决现有 的利用常规35s为启动子的crispr/cas9基因编辑载体进行编辑时,后代植株 出现嵌合的比例较高且编辑效率低,耗费大量的人力、物力、财力的问题。
6.本发明通过构建烟草基因编辑载体时,以生殖细胞特异高表达ntec1.2
‑
1 基因启动子驱动cas9蛋白表达,,实现降低烟草嵌合体的比率,提高基因编辑 效率的目的。
7.本发明是通过以下技术方案实现的:
8.一种烟草生殖细胞特异高表达基因,核苷酸序列如seq id no.1所示, 包括372个碱基,来源于烟草,命名为ntec1.2
‑
1。
9.优选的,所述烟草生殖细胞特异高表达基因的启动子,核苷酸序列如 seq id no.2所示,包括2186个碱基。
10.优选的,引物的核苷酸序列如seq id no.3和seq id no.4所示。
11.基因编辑烟草生殖细胞特异高表达基因,获得具有特异性表达的烟草植 株。
12.优选的,烟草生殖细胞特异高表达基因的编码序列设计特异定量引物,分 别提取盛花期烟草的各组织的总rna,通过反转录获得cdna,并以其为模板, 利用荧光定量pcr的方法检测基因在烟草各组织的表达情况,获得的数据利用 2
‑
δδct的方法分析基因在烟草各组织的表达情况,确定该基因是在生殖器 官中特异高量表达。
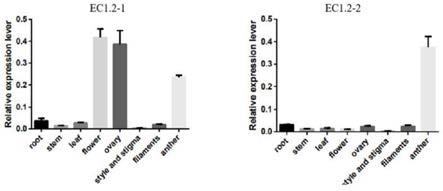
13.优选的,编辑烟草生殖细胞特异高表达基因所需的特异定量引物的核 苷酸序列如seq id no.5和seq id no.6所示。
14.优选的,基因编辑烟草生殖细胞特异高表达基因的启动子,获得具有 可在烟草生殖细胞特异高表达的编辑载体优选的,通过含pore
‑
ntec1.2
‑
1p: gus载体的农杆菌侵染烟草植株植,表明ntec1.2
‑
1启动子在早期的种子和子 房中是特异表达的。。
15.优选的,将含有pore
‑
ntec1.2
‑1‑
rbcs
‑
e9敲除载体的农杆菌侵 染烟草植株,获得转基因烟草植株,经突变检测显示在检测的15株 转基因品系中有4株发生了突变,突变效率为26.7%。
16.本发明的有益效果是:
17.本发明通过基因组信息学分析,鉴定获得栽培烟草ntec1.2
‑
1基因的编码 序列及其启动子序列。通过候选启动子序列元件分析及基因表达谱分析,证实 ntec1.2
‑
1是一个生殖细胞高丰度表达的基因。克隆获得ntec1.2
‑
1的启动子 序列,并以其为启动元件构建生殖细胞特异高丰度表达cas9蛋白的基因编辑 载体ntec1.2
‑1‑
cas9
‑
rbcs
‑
e9。载体转入农杆菌,通过叶盘转化法获得转基 因烟草植株。通过基因编辑靶位点测序分析发现,转基因烟草植株pds基因突 变效率高达26.7%。研究结果对于构建高效率的植物基因失活载体并获得突变 植株具有重要的指导意义及应用价值。
附图说明
18.图1为ntec1.2基因在烟草不同组织的表达情况示意图;
19.图2为烟草ntec1.2
‑
1基因启动子的克隆电泳图,其中a为ntec1.2
‑
1 基因启动子序列pcr产物电泳图,b为拟南芥atec1.2基因启动子序列pcr产 物电泳图,p表示启动子;
20.图3为启动子活性验证荧光显微镜图;
21.图4为启动子载体构建电泳图,其中,a为nhei和noti双酶切gus载体, 1:酶切后gus载体;2:原始gus载体;b为nhei和noti双酶切启动子序列; 1,2:ntec1.2
‑
1启动子;3,4:对照启动子;
22.图5为gus转基因植株pcr鉴定电泳图,其中1
‑
12为不同植株编号;
23.图6为转基因gus幼苗染色图片;
24.图7为转基因组织gus染色图片;
25.图8为特异性启动子敲除载体构建电泳图,其中,a:1,2为rbcs
‑
e9终 止子;b:1表
示ncoi和noti酶切后的pore
‑
cas9载体,2表示pore
‑
cas9 原始载体,4表示nhei和ecori酶切后pore
‑
cas9
‑
rbcs
‑
e9载体,5表示
26.pore
‑
cas9
‑
rbcs
‑
e9原始载体。
具体实施方式
27.以下通过实施例来详细说明本发明技术方案,以下的实施例仅是示例性 的,仅能用来解释和说明本发明的技术方案,而不能解释为是对本发明技术方 案的限制。
28.在本技术的各实施例中,没有注明具体技术或条件者,按照本领域内现有 技术或条件进行,所使用的材料或设备未注明生产厂商者,均为可以通过购买 获得的常规产品。
29.本发明除非另有说明,否则百分号为体积百分数,比例为体积比。
30.本技术中所使用的烟草品种为红花大金元,一种商品化烟草品种。
31.本技术提供一种烟草生殖细胞特异高表达基因,从ncbi中得到的拟南芥 的atec1.2基因的氨基酸序列,将其比对烟草基因组数据库,在烟草中获得了 atec1.2的两个同源候选基因,将其命名为ntec1.2
‑
1和ntec1.2
‑
2。其中 ntec1.2
‑
1的核苷酸序号如seq id no.1所示,包括372个碱基。
32.利用genomatix在线分析网站对所比对到的ntec1.2
‑
1和ntec1.2
‑
2基因 上游2200bp序列进行活性及特异性的元件分析。发现这两个基因的启动子上 除了含有启动子核心元件tata盒和caat盒外,还有一些特有的元件: ntec1.2
‑
1p和ntec1.2
‑
2p分别含有15和9个pollen ilat52(agaaa)元件,5 个和7个花粉特异性转录enhance element(tgtga),6个和3个细胞分裂m 时期特异性元件(aacgg)。ntec1.2
‑
1p含有2个早期激活因子的靶点 (aacttaa),这些都是参与生殖或早期生长发育相关的特异性元件,这些元件 的鉴定表明了我们所得到的序列有可能是生殖特异性的启动子,得到烟草生殖 细胞特异高表达基因的启动子,核苷酸序列如seq id no.2所示,包括 2186个碱基。
33.为了进一步证明获得的ntec1.2
‑
1和ntec1.2
‑
2基因是在生殖器官中特异 表达,对其在烟草各组织的相对表达情况进行了定量分析。
34.首先根据ntec1.2
‑
1和ntec1.2
‑
2基因的编码序列设计特异定量引物,分 别提取盛花期烟草的子房、花柱、花丝、花药、花、根、茎、叶的总rna,通 过反转录获得cdna,并以其为模板,利用荧光定量pcr的方法检测这两个基 因在烟草8个组织的表达情况。获得的数据利用2
‑
δδct的方法分析这两个 基因在烟草八个组织的表达情况。处理后的结果利用graphpad prism7软件作 图进行绘图分析。结果如图1所示,ntec1.2
‑
1和ntec1.2
‑
2基因均在花药中 高量表达,其中ntec1.2
‑
2相较于各组织特异高量表达,而ntec1.2
‑
1不但在 花药中高量表达,在子房中也是高量表达的,该结果证明了以上两个基因是在 生殖器官中特异高量表达,可能参与植物的生殖和早期发育。
35.具体的ntec1.2基因表达特征分析:
36.总rna的提取
37.取3株盛花期的烟草,分别对其子房、花柱、花丝、花药、花、根、茎、 叶8个组织进行取材,把三株相同组织的材料放入离心管中后迅速的放入液氮 中,利用easypure plant rna kit试剂盒(来自全式金公司)进行rna提取 (以下操作均在室温下进行)。
38.(1)将干热灭菌的研钵和研棒倒入液氮进行预冷,将取的烟草各组织样 品分别放
入研钵中。
39.(2)加入液氮并快速的研磨,待液氮即将完全蒸发时立即加入液氮,并 继续研磨,重复操作3
‑
4次,直至样品成粉末状。把样品收集到1.5ml无rna 酶离心管中。
40.(3)加入5μlβ
‑
巯基乙醇和500μl bb6,震荡混匀后静置3min。
41.(4)12000
×
g离心5min,吸取上清到新的1.5ml无rna酶离心管中, 加入0.5倍体积的无水乙醇,翻转混匀。
42.(5)吸取混合液于离心柱中,12000g离心30s,弃流穿液。
43.(6)吸取500μl cb6于离心柱中,12000g离心30s,弃流穿液,加入 80μl dnasei并静置15min,继续加500μl cb6,12000g离心30s,弃流 穿液。
44.(7)吸取500μl wb6于离心柱中,12000g离心30s,弃流穿液。
45.(8)重复该步骤(7)一次。
46.(8)12000g离心2min,静置2min使离心柱中残留的乙醇挥发。
47.(9)吸取30μl无rna酶水悬空加入离心柱中,静置1min后12000g 离心2min。将洗脱的rna放置在
‑
80℃冰箱保存。
48.cdna的获得:
49.(1)按照如下体系加样,然后放入pcr仪中70℃,5min,冰浴2min。
[0050][0051]
(2)然后向体系中加入如下试剂并混匀,置于pcr仪中42℃,60min, 4℃保存。
[0052][0053]
荧光定量pcr检测基因表达水平:
[0054]
(1)使用miscript sybr green pcr kit(来自qiagen公司)按如下体 系进行加样,每个样品进行三次技术重复,混匀离心后按如下扩增程序进行。
[0055][0056][0057]
(2)对扩增结束的程序结果进行溶解曲线分析,然后利用2
‑
δδct
的方法 对数据进行处理并计算出基因的相对表达量。将计算出的结果进行作图,分析 基因在各组织中的表达情况。
[0058]
烟草生殖特异性基因ec1.2的组织特异性表达验证方法:
[0059]
将本氏烟草种子均匀的撒在湿润的土壤(营养土:蛭石=2:1)表面,并用 有透气孔的保鲜膜封住盆口,26℃,光照16h,黑暗8h,湿度70%,培养 一周,期间保持土壤湿润,待种子出芽一周后将其移栽到适当的盆中继续培养。
[0060]
亚细胞定位载体构建:
[0061]
)pbi221
‑
eefp载体是购自重庆金喜鹊公司。
[0062]
(2)用neb公司的sali
‑
hf和ncoi
‑
hf双酶切pbi221
‑
eefp植物表达载 体。通过切胶回收的方式回收pbi221
‑
eefp骨架载体。
[0063]
(3)分别设计含有sali
‑
hf的正向特异性引物和ncoi
‑
hf的反向特异性 引物扩增启动子序列。
[0064]
(4)利用neb公司的gibson assembly mastermix酶按照如下体系加样, 置于pcr仪中50℃孵育30min。
[0065][0066][0067]
(5)将孵育后的产物直接转化到dh5
‑
α大肠杆菌中,37℃过夜倒置培 养。
[0068]
(6)挑取单克隆利用特异性引物进行pcr扩增,通过琼脂糖凝胶电泳, 进行初步检测,将阳性克隆的菌液送公司进行sanger测序。
[0069]
(7)提取测序结果正确的菌液的质粒,放置
‑
20℃冰箱保存备用。
[0070]
gv3101农杆菌介导的瞬时表达:
[0071]
从
‑
80℃冰箱取出gv3101农杆菌感受态细胞,置于冰面冻融。取0.5
‑
1μg 质粒加入到含有50μl感受态细胞的离心管中,按照冰浴5min、液氮冷冻5 min、37℃水浴热激5min、冰浴5min的先后顺序进行。
[0072]
(2)加入500μl室温的sod液体培养基,放置在恒温摇床上,28℃,200 g,振荡培养2
‑
3h后6000g离心2min,弃上清后加入100μl的sod液体 培养基,悬浮菌体后均匀涂布在含有50mg/l卡那霉素、20mg/l利福平、40 mg/l庆大霉素的yeb平板上,放于28℃恒温培养箱倒置避光培养2天。
[0073]
(3)设计特异性的引物对平板中的单菌落进行pcr检测,将阳性克隆置 于含有三抗的液体yeb培养基中,扩大培养待菌液浑浊后,取1ml菌液接种 至50mlyeb培养基中进一步扩大培养10h后,4℃,3000g离心15分钟 收集菌体。
[0074]
(4)将收集到的菌液用ms液体培养基悬浮,使其od值调至0.6
‑
0.8。 放置室温中避光黑暗静置2
‑
3h。
[0075]
(5)使用生长约30d左右的有宽大嫩绿叶片的本氏烟草用于注射。
[0076]
(6)用除去针头的1ml注射器,选取嫩绿宽大的叶片从叶背缓缓注射菌 液直至菌液侵染整个叶片。
[0077]
(7)将注射的本氏烟草避光培养8h,随后在光照16h,黑暗8h的环 境中培养,培养36h
‑
48h后可进行荧光观察。
[0078]
gus植物表达载体的构建:
[0079]
)pore
‑
gus载体是本实验保存。
[0080]
(2)用neb公司的nhei
‑
hf和noti
‑
hf双酶切pore
‑
gus植物表达载体, 并通过切胶回收的方式回收骨架载体。
[0081]
(3)分别设计含有骨架载体切口同源臂的特异性引物扩增启动子序列。
[0082]
(4)利用neb公司的gibson assembly master mix酶按照如下体系加样, 置于pcr仪中50℃孵育30min。
[0083][0084]
(5)将孵育后的产物直接转化到dh5
‑
α大肠杆菌中,37℃过夜倒置培 养。
[0085]
(6)挑取单克隆利用特异性引物进行pcr扩增,通过琼脂糖凝胶电泳, 进行初步检测,将阳性克隆的菌液送公司进行sanger测序。
[0086]
(7)提取测序结果正确的菌液的质粒,放置
‑
20℃冰箱保存备用。
[0087]
稳定遗传的gus转基因植株的获得:
[0088]
将构建好的质粒转化到lba4404农杆菌中,扩大培养后用ms液体培养基 悬浮菌液备用。
[0089]
(2)打叶盘:将生长在培养瓶中的四叶期的无菌秒在灭菌后的超净工作 台中去除置于滤纸上,用规格为5mm
×
5mm打孔器选取嫩绿叶片进行打孔, 避开叶脉,将获得叶盘收集,50个叶盘为一组,放置入含有ms液体培养基的 瓶子中备用。
[0090]
(3)农杆菌重悬液侵染叶盘:将叶盘捞出放入新的培养瓶中,加入含有 lba4404农杆菌的重悬液,使叶盘完全浸在重悬液中,侵染8min,将叶盘捞 出放置在滤纸上吸干液体,然后用镊子将叶盘放在ms固体培养基上,28℃黑 暗培养两天。
[0091]
(4)诱导愈伤组织的生成:将叶盘转移至含有植物激素,卡那霉素,羧 苄霉素
[0092]
的选择培养基上。在28℃,光照16h,黑暗8h的温室中继续培养,期 间每隔14d更换一次选择培养基,直至叶盘分化出愈伤组织细胞团。
[0093]
(5)愈伤组织分化成芽:将形成的愈伤组织分散开置于新的选择培养基 中继续培养,两周后会从愈伤组织中分化出幼芽。
[0094]
(6)诱导生根:用无菌手术刀将生长在愈伤组织上的幼芽切下,插入生 根培养基中,培养10
‑
14d后生根,至此,我们获得了无菌的再生烟草植株。
[0095]
gus转基因植株鉴定:
[0096]
提取转基因gus植株的基因组,设计特异性引物对gus基因进行pcr扩增, 获得的pcr产物利用全式金公司的pcr产物纯化试剂盒进行纯化(详细步骤见 试剂盒说明书),送
测序公司进行sanger测序,将测序结果与gus基因序列进 行比对,成功比对的pcr产物对应的植株即为gus转基因植株。pcr扩增体系 和程序如下:
[0097][0098][0099][0100]
gus染液的配置:
[0101]
x-gluc母液:x
‑
gluc(5
‑
溴
‑4‑
氯
‑3‑
吲哚
‑
β
‑
葡萄糖苷),用n
‑
n
‑
二甲 基酰胺(dmf)配成20mm的贮存液,分装成每管100μl,保存于
‑
20℃。
[0102]
(2)x-gluc基液,配制方法:
[0103][0104]
gus染色液:50μl x
‑
gluc母液+50μl基液。
[0105]
gus植株染色:
[0106]
(1)取gus转基因植株的幼苗和盛花期的花苞、花、花药、子房(同时 取相同生长状态的含有pore
‑
gus空载质粒的植株作对照)分别放入含有gus 染色液的试管中,通过真空渗入处理,保证组织完全浸泡在染色液中,放入 37℃恒温培养箱中,过夜孵育。
[0107]
(2)将染色后的组织放入含有70%乙醇中进行脱色处理,期间更换乙醇 2
‑
3次,直至我们的对照组材料呈白色。
[0108]
(3)通过显微镜或者肉眼直接观察,于对照组比较各组织部位的染色情 况。
[0109]
ntec1.2启动子的克隆:
[0110]
为了获得以上两个基因的启动子序列,首先根据ntec1.2
‑
1和ntec1.2
‑
2 基因启动子的预测序列,分别对其设计特异性引物,以烟草花粉组织cdna为 模板,分别对两条启动子序列进行pcr扩增。同时也对拟南芥的atec1.2基因 的启动子为模板进行pcr扩增。琼脂糖凝胶电泳结果显示(图2),以上3个 基因的启动子条带大小与预测序列的长度相符合,其中pro:ntec1.2
‑
1和 pro:ntec1.2
‑
2长度分别为2118bp和2201bp。将pcr产物通过切胶回收连 接到ta载体上,转化到dh
‑
5α大肠杆菌,通过卡那霉素抗性筛选后挑取单克 隆进行菌落pcr初步检测,将阳性单克隆菌液送公司测序,测序结果与获得的 数据库序列进行blast比对,序列是一致的。将正确的菌液提取质粒于
‑
20℃ 冰箱储存。
[0111]
具体步骤为:
[0112]
(1)选取野生型的嫩绿烟草叶片约100mg,加入液氮在研钵中充分研磨, 放入1.5ml离心管中并加入250μl rb1和15μl rnase a,震荡使其充分混 匀。放入55℃水浴锅中孵育15min。12000g离心15min,吸取上清于新的 离心管中,加入100μl pb1后充分混匀,冰浴5min,12000g离心5min。 吸取上清于干净的离心管中加入375μl含有无水乙醇的bb1混匀后加入到离 心柱中,12000g离心30s,弃流出液。然后加入500μl cb1,12000g离心 30s,弃流出液。加入500μl wb1,12000g离心30s,弃流出液。然后12000 g空离2min,将离心柱放入新的1.5ml的离心管中,室温放置一分钟,加入 65℃的ddh2o室温放置一分钟后,12000g离心1min,洗脱在离心管底部的 液体即为烟草基因组。
[0113]
(2)设计特异性性引物,利用高保真酶对预测启动子进行pcr扩增,扩 增体系和程序如下:
[0114]
[0115][0116]
(2)将pcr产物割胶回收后连接到peasy
‑
t5载体上,将载体转化到 dh
‑
5α大肠杆菌中,37℃复苏40min后涂布到含有卡那霉素的lb平板中, 37℃倒置避光过夜培养。
[0117]
(3)从平板中挑取单克隆,利用m13f/r通用引物进行菌液pcr,扩增体 系和程序如下:
[0118][0119][0120]
(4)将pcr产物进行琼脂糖凝胶电泳,将条带大小与克隆的启动子序列 大小相吻合的菌液或pcr产物送测序公司进行sanger测序。测序结果与数据 库中获得的启动子序列进行blast比对,将比对结果完全正确的菌液,扩大培 养,进行质粒的提取,然后放置在
‑
20℃冰箱保存备用。至此,得到了烟草内 源的ec1.2启动子。
[0121]
构建含有生殖特异性启动子的egfp植物表达载体:
[0122]
通过设计了含有sali酶切位点及骨架载体同源臂的正向引物和含有ncoi 酶切位点及骨架载体同源臂的反向引物特异性扩增启动子序列,使用sali
‑
hf 和ncoi
‑
hf双酶切pbi221
‑
eefp植物表达载体并回收其骨架载体。采用无缝克 隆的原理,用neb公司的gibson assembly master mix酶,将启动子构建到 pbi221
‑
egfp载体上。
[0123]
为了探究克隆的启动子是否有活性,将构建好的egfp植物表达载体 pbi221
‑
ntec1.2
‑1‑
egfp、pbi221
‑
ntec1.2
‑2‑
egfp和pbi221
‑
35s
‑
egfp通过 化学转化法转入gv3101农杆菌中。将阳性农杆菌菌落扩大培养后,用ms液体 培养基调节菌液od值到0.6
‑
0.8,黑暗静置2
‑
3h后,用1ml注射器吸取适 当的菌液从本氏烟草的叶背进行缓缓注射,直至整个叶片完全浸染菌液。为了 获得更可靠的结果,将每种菌液在一株本氏烟草的三片叶子进行注射,并用标 签进行标记,注射完成后将本氏烟草置于黑暗环境培养8h,然后进行正常培 养。侵染36
‑
48h后,取不同叶片置于荧光显微镜下进行观察,结果表明,与 对照组(35s:egfp)相比,2个实验组全都有绿色荧光信号的出现。实验组 绿色荧光的出现说明了egfp荧光蛋白有所表达,证明了克隆的2个生殖特异 性启动子是有活性的,具体见图3。
[0124]
为了验证克隆的启动子不仅有活性,而且是特异性表达的,构建了 pore
‑
ntec1.2
‑
1p:gus和pore
‑
ntec1.2
‑
2p:gus植物表达载体。首先分别设计 特异引物克隆启动子得到含有nhei酶切位点和noti酶切位点的ntec1.2
‑
1p 和ntec1.2
‑
2p序列(图4),用neb公司的nhei
‑
hf酶和noti
‑
hf酶分别对 pore
‑
gus植物表达载体和克隆的启动子进行双酶切(图4),分别进行切胶 回收,将载体骨架(pore
‑
gus)和片段(ntec1.2
‑
1p和ntec1.2
‑
2p)通过t4 连接酶进行连接。连接产物用化学转化法转化到大肠杆菌感受态中,用含有卡 那霉素的lb平板培养后,挑取单克隆菌落,用特异引物进行检测,根据琼脂 糖凝胶电泳结果,选择可能的阳性菌落进行测序,将测序结果正确的菌液提取 其质粒。放置
‑
20℃冰箱储存备用。
[0125]
pore
‑
gus转基因烟草植株的获取:将构建好的pore
‑
ntec1.2
‑
1p:gus和 pore
‑
ntec1.2
‑
2p:gus植物表达载体分别转化到lba4404农杆菌中,通过抗 性培养基筛选培养后,挑取阳性单克隆扩大培养,用ms液体培养基将菌液悬 浮,并使其od值达到0.6
‑
0.8,室温黑暗静置2
‑
3h后,倒入含有叶盘的玻 璃瓶中,使其叶盘完全浸没叶盘,侵染8min后,用滤纸吸干液体,将叶盘放 入ms固体培养基中共培养2d后,转入到含有卡那霉素和羧苄霉素的培养基 中进行选择培养,期间更换培养基,直至叶盘脱分化形成愈伤组织,对愈伤组 织进行修剪后继续培养,直至愈伤组织分化成芽,将芽切下放入生根培养基中 培养直至生根。提取生根植株叶片基因组,用特异性引物进行pcr扩增gus 基因序列,琼脂糖凝胶电泳初步检测后,送公司进行测序,测序结果与数据库 序列进行blast比对,比对正确的植株即为转基因gus烟草植株(图5)。
[0126]
取pore
‑
ntec1.2
‑
1p:gus、pore
‑
ntec1.2
‑
2p:gus植株的幼苗和盛花期 的花苞、花、花药、子房(同时取相同生长状态的野生型植株作为对照)分别 放入配置好的gus染色液中,由于烟草的组织较厚不易染色,我们将花苞纵向 切割成两半,用去掉针头的针管抽取试管中的空气,使染色液完全渗入烟草组 织中,放入37℃恒温培养箱中,过夜孵育。用75%的无水乙醇对孵育后的各 组织进行脱色处理,脱色2
‑
3次,直至对照组染色完全脱去。然后分别用显微 镜观察和肉眼直接观察。发现与对照组相比,pore
‑
ntec1.2
‑
1p:gus和 pore
‑
ntec1.2
‑
2p:gus植株转基因植株的幼苗都是呈现白色。该结果说明gus 基因在幼苗
中没有表达或者表达量极低,进一步表明了ntec1.2
‑
1p和 ntec1.2
‑
2p启动子在幼苗期是没有表达的(图6)。而在花苞和种子的染色中, pore
‑
ntec
‑
1.2
‑
1p:gus实验组植株的种子和花苞剖面的子房处显现出明显的 蓝色,而pore
‑
ntec1.2
‑
2p:gus实验组在种子和花苞子房处没有显色或在种 子叶柄处有很浅的蓝色出现,该结果说明了ntec1.2
‑
1启动子在早期的种子和 子房中是特异表达的,而且从染色的程度看,表达量也很高。而 pore
‑
ntec1.2
‑
2p:gus植株在早期种子和子房处没有明显的染色出现,说明ntec1.2
‑
2启动子在早期种子时期和子房处没有表达或者在其他时期表达(图 7)。
[0127]
特异性启动子敲除载体的构建及编辑效率检测:
[0128]
首先,以含有ntec1.2
‑
1和ntec1.2
‑
2启动子序列的t5载体为模板,用 特异性引物分别扩增ntec1.2
‑
1、ntec1.2
‑
2启动子序列。然后,用特异性引 物扩增公司合成的rbcs
‑
e9终止子片段。利用ncoi和noti酶切pore
‑
cas9 载体,用t4连接酶将载体中原有的nos终止子替换成我们克隆的rbcs
‑
e9终 止子。通过nhei和ecori酶切pore
‑
cas9
‑
rbcs
‑
e9载体,利用t4连接酶将原 有的35s启动子替换成克隆的ntec1.2
‑
1和ntec1.2
‑
2启动子,以驱动cas9 的表达。至此,pore
‑
ntec1.2
‑1‑
rbcs
‑
e9、pore
‑
ntec1.2
‑2‑
rbcs
‑
e9敲除载 体构建完成。同时,用同样的方法构建了pore
‑
atec1.2
‑
rbcs
‑
e9载体,作为 对照质粒,结构示意图如下图8。
[0129]
将构建好的pore
‑
ntec1.2
‑1‑
rbcs
‑
e9、pore
‑
ntec1.2
‑2‑
rbcs
‑
e9以及对 照质粒pore
‑
atec1.2
‑
rbcs
‑
e9利用农杆菌介导的遗传转化和组织培养技术获 得t0代转基因植株。此处用的为含有利福平(rif)、链霉素(str)和卡那霉 素(kan)的yeb抗性培养基。通过打叶盘、农杆菌侵染、农杆菌共培养、选 择培养、生根培养等农杆菌介导的遗传转化和组织培养获得t0代转基因植株。 用特异性jc
‑
f对植株进行阳性检测。每种转化敲除载体获得的植株为一个品 系,三种品系每种检测20株转基因阳性植株。
[0130]
t1代转基因植株的获得:
[0131]
将收取到的3组转基因烟草(pore
‑
ntec1.2
‑1‑
rbcs
‑
e9、 pore
‑
ntec1.2
‑2‑
rbcs
‑
e9、pore
‑
atec1.2
‑
rbcs
‑
e9)的t0代种子用75%的酒 精浸泡t0代转基因植株的种子30s后,用灭菌的ddh2o洗涤2
‑
3次,再用5% 次氯酸钠浸泡8min,期间轻容震荡2
‑
3次后用灭菌的ddh2o洗涤2
‑
3次,倒 于滤纸中吸干水分等步骤进行消毒灭菌处理后,将其放在含有卡那霉素的ms 固体培养基上,用封口膜将平板密封,放置在26℃的温室中,光照16h,黑 暗10h。约7d后种子萌发长出幼芽,待幼芽长到1
‑
2cm时,将其分别移栽 到含有卡那霉素的ms固体培养基的大玻璃瓶中继续培养。通过抗性筛选后, 每组植株随机挑选,提取其基因组,并通过特异性引物进行pcr扩增载体sgrnascaffold序列,琼脂糖凝胶电泳初步鉴定后,将阳性菌落送公司进行测序, 将测序结果进行blast比对,筛选出每组烟草的转基因植株。检测了 pore
‑
ntec1.2
‑1‑
rbcs
‑
e9、pore
‑
ntec1.2
‑2‑
rbcs
‑
e9品系各20株,检测到 的转基因植株分别为16株和19株,转基因阳性率分别为80%和95%。检测 了pore
‑
atec1.2
‑
rbcs
‑
e9品系25株,检测到的转基因植株为21株,转基因 阳性率为84%。
[0132]
t1代转基因植株突变检测:
[0133]
分别提取检测结果为阳性的转基因株系的基因组,用检测引物jc
‑
f和 u26
‑
r检测叶片基因组中的sgrna及grna scaffold基因片段鉴定烟草是否为 转基因植株。用正向引物5'ctgaagcagtcaccaagaat 3'和反向引物5' agctatctgattaatggacaaa 3'扩增烟草pds
基因序列,进行pcr扩增,将pcr 产物切胶回收后连接到t载体上,转化涂板后,挑取单克隆,进行琼脂糖凝胶 电泳初步检测,将条带大小与扩增片段相吻合的样品送公司进行测序检测。检 测结果与pds基因序列进行blast比对,结果发现在检测的20株 pore
‑
atec1.2
‑
rbcs
‑
e9转基因品系中有4株发生了突变,突变效率为20%, 突变形式均为碱基的缺失。检测的15株pore
‑
ntec1.2
‑1‑
rbcs
‑
e9转基因品系 中有4株发生了突变,突变效率为26.7%,突变形式为碱基的缺失和插入。 检测的20株pore
‑
ntec1.2
‑2‑
rbcs
‑
e9转基因品系中有1株发生了突变,突变 效率为5%,突变形式为碱基的替换,突变信息见表1。
[0134]
表1t1代转基因植株突变信息统计表
[0135][0136][0137]
其中加深字母代表pam区,斜体字母和点代表碱基的缺失、插入和替换。 以上显示和描述了本发明的基本原理、主要特征和本发明的优点。
[0138]
本行业的技术人员应该了解,本发明不受上述实施例的限制, 上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本 发明精神和范围的前提下,本发明还会有各种变化和改进,这些变 化和改进都落入要求保护的本发明范围内。本发明要求保护范围由 所附的权利要求书及其等效物界定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1