恩格列净中间体的制备方法与流程
1.本发明属于医药化工领域,具体涉及恩格列净及其中间体的制备方法。
背景技术:
2.恩格列净(empagliflozin)是由勃林格殷格翰和礼来公司联合开发的一种选择性2型钠葡萄糖协同转运蛋白(sglt2)抑制剂,已临床用于治疗2型糖尿病,并已在美国、欧洲、日本、中国等多个国家上市销售。恩格列净是含(s)-3-芳氧基取代四氢呋喃和-d-葡萄糖醇结构片段的手性化合物。两个手性片段主要是通过以(s)-3-(4-(5-溴/碘-2-氯苄基)苯氧基)四氢呋喃为关键中间体与葡萄糖苷內酯经亲核加成反应(j.label compd.radiopharm.2014,57,687-694;org.lett.2014,16,4090-4093)或钯催化偶联的方法(org.lett.2018,20,1936-1940)对接完成恩格列净的合成。其中,关键中间体(s)-3-(4-(5-溴/碘-2-氯苄基)苯氧基)四氢呋喃中手性四氢呋喃片段的引入也主要是通过(s)-或(r)-3-羟基四氢呋喃为手性原料经mitsunobu亲核取代反应等来合成,存在原子经济性差等缺点。此外,还存在原料成本高、后处理繁琐、环境友好性差等不足。然而,目前大部分国内外专利和文献均采用这样的传统合成方法来合成。
3.此外,不对称催化是合成手性化合物最有效的方法,特别是具有原子经济好,绿色、高效、环境友好特征的不对称催化氢化已在手性药物等的工业生产中得到了广泛的应用。但由于缺少相应的高效不对称催化氢化等不对称合成方法和技术,目前仍没有用不对称催化合成这一恩格列净关键手性中间体的报道。因此,针对现有合成恩格列净关键手性中间体方法及技术上的缺点和不足,本发明发展并提供了一种通过α-芳氧基取代γ-內酯的不对称催化氢化为关键步骤不对称合成恩格列净关键手性中间体的新方法。从简单的原料出发,只需包括不对称催化氢化在内的4步反应就可以完成(s)-3-(4-(5-溴/碘-2-氯苄基)苯氧基)四氢呋喃的不对称合成。该合成新方法操作简单,避免了原子经济性差的mitsunobu亲核取代反应,具有绿色、高效、环境友好等优点。
技术实现要素:
4.本发明提供了一种恩格列净的中间体及其制备工艺,该方法操作简单,成本低廉,原子经济性好,适于工业化生产。
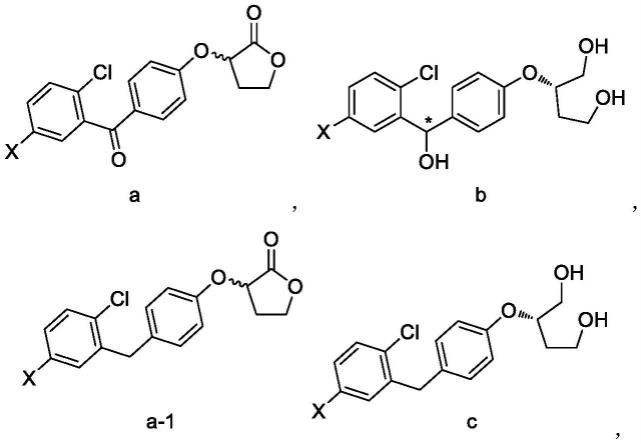
5.首先,本发明提供了通式结构的式a-1或式a-2化合物,
[0006][0007]
其中,x为卤素,较优选地为溴或碘,r1,r2相同的为羟基或组合成内酯环,r3为氢或
羟基。
[0008]
进一步,本发明提供了下述结构的化合物,
[0009][0010]
其中,x为卤素,较优选地为溴或碘。
[0011]
本发明提供了式a化合物的制备方法,由化合物2与化合物3经傅克酰基化反应制备。
[0012]
所述傅克酰基化反应路线如下:
[0013][0014]
其中x为卤素,较优选地为溴或碘。
[0015]
所述傅克酰基化反应中反应温度范围为50-150℃,所述催化剂为多聚磷酸(ppa)等。
[0016]
本发明提供了式a-1化合物的制备方法,由式a化合物在还原剂作用下经还原反应制备得到,
[0017][0018]
其中x为卤素,较优选地为溴或碘。
[0019]
所述还原反应中还原剂可以为硼氢化钠、硼氢化钾、三乙基硅烷、1,1,3,3-四甲基二硅氧烷或四甲基二硅氮烷等;所述还原反应需要添加质子酸或酸如氯化铝、三氟乙酸等;所述反应溶剂为二氯甲烷、乙腈、甲苯、四氢呋喃、二氧六环、n,n-二甲基甲酰胺、二甲亚砜
中的一种或其中几种的混合溶剂;所述反应温度范围为-1085℃。
[0020]
本发明提供了式b化合物的制备方法,由式a化合物经不对称催化氢化反应制备,
[0021][0022]
其中,x为卤素,较优选地为溴或碘。
[0023]
本发明提供了式c化合物的制备方法,由式b化合物经脱苄位羟基反应制备,或由式a-1化合物经不对称催化氢化反应制备,
[0024][0025][0026]
其中,x为卤素,较优选地为溴或碘。
[0027]
上述不对称催化氢化反应中,所述催化剂为手性催化剂,通式结构为式5化合物或式6化合物。
[0028][0029]
其中,式5化合物中n为0-4;r1选自c1-c10的烃基、苯基、取代苯基、1-萘基、2-萘基、杂芳基或苄基,所述的苯基上的取代基为c1-c10的烃基、烷氧基,取代基数量为1-5,杂芳基为呋喃基、噻吩基或吡啶基;
[0030]
式6化合物中r1、r2选自c1-c10的烃基、苯基、取代苯基、1-萘基、2-萘基、杂芳基或苄基,所述苯基上的取代基为c1-c10的烃基、烷氧基,取代基数量为1-5,杂芳基为呋喃基、噻吩基或吡啶基;其中,r1、r2可以相同也可以不同;
[0031]
式5和6化合物的典型代表如下:
[0032][0033]
所述不对称氢化反应中,手性催化剂与化合物a或a-1的摩尔比为100:1-50000:1;化合物a或a-1浓度为0.001-10.0m;
[0034]
所述不对称氢化反应在碱的存在条件下进行。所述碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、乙醇钠、乙醇钾、叔丁醇钠、叔丁醇钾、叔丁醇锂、三乙胺、三丁胺或n-甲基吗啉;所述碱浓度为0.005m-1.0m。所述氢气压力为0.1-100atm;所述反应温度范围为0-80℃;所述有机溶剂为甲醇、乙醇、丙醇、异丙醇、丁醇、四氢呋喃、甲苯、甲基叔丁基醚、二氧六环、n,n-二甲基甲酰胺、二甲亚砜中的一种或其中几种的混合溶剂。
[0035]
本发明提供了恩格列净中间体式d化合物的制备方法,由式c化合物经脱水关环反应制备,
[0036][0037]
其中,x为卤素,较优选地为溴或碘;
[0038]
所用脱水剂为对甲苯磺酸、对甲苯磺酰氯、苯磺酸、硫酸、甲基磺酰氯;反应溶剂为二氯甲烷、乙腈、甲苯、四氢呋喃、二氧六环、n,n-二甲基甲酰胺、二甲亚砜中的一种或其中几种的混合溶剂;所述反应温度为20-150℃。
[0039]
本发明提供的恩格列净中间体的合成是通过不对称催化氢化为关键步骤来合成,具有操作简单,原子经济好,收率高等优点,这为手性药物恩格列净的关键手性中间体的不对称合成提供了绿色、高效、环境友好的合成新方法。
具体实施方式
[0040]
为了进一步理解本发明,下面结合实施例对本发明提供的恩格列净中间体的制备方法进行详细说明。需要理解的是,这些实施例描述只是为进一步详细说明本发明的特征,而不是对本发明范围或本发明权利要求范围的限制。
[0041]
实施例1:
[0042][0043]
250ml两口瓶中加入多聚磷酸(37.6g),升温至60℃,随后依次加入5-溴-2氯苯甲酸式3化合物(4.72g,20mmol)和化合物2(3.56g,20mmol)。升温至100℃反应4小时。随后冷却至50℃并加入水(100ml)。继续搅拌1小时,加入二氯甲烷(50ml),继续搅拌1小时后,分出有机相,水相用二氯甲烷萃取(30
×
3ml)。合并有机相,有机相经饱和食盐水洗涤,无水硫酸镁干燥。减压脱除溶剂得到粗品,柱层析(石油醚/乙酸乙酯(4:1-2:1))得到化合物a黄色固体4.5g,收率58%。熔点:102-105℃.1h nmr(400mhz,cdcl3)δ7.86
–
7.74(m,2h),7.55(dd,j=8.6,2.4hz,1h),7.48(d,j=2.4hz,1h),7.33(d,j=8.4hz,1h),7.16
–
7.01(m,2h),5.08(t,j=8.0hz,1h),4.61
–
4.50(m,1h),4.45
–
4.35(m,1h),2.84
–
2.71(m,1h),2.58
–
2.44(m,1h).
13
c nmr(101mhz,cdcl3)δ192.0,172.6,161.7,140.3,133.9,132.5,131.6,131.5,130.2,130.1,120.6,115.6,72.1,65.4,29.7.hrms(esi)calcd for c
17h13
brclo4([m+h]
+
):394.9680;found:394.9681.
[0044]
实施例2:
[0045][0046]
250ml两口瓶中加入alcl3(2.4g,18.2mmol),加入化合物a(3.6g,9.1mmol)。置换氩气,加入甲苯(100ml)。体系冷却至0℃,缓慢加入1,1,3,3-四甲基二硅氧烷(2.43g,18.2mmol),随后恢复室温搅拌1小时。反应结束后冷却至0℃,加入冰水淬灭,分液,水相用乙酸乙酯(20ml
×
3)萃取,合并有机相,无水硫酸镁干燥,抽滤,减压脱除溶剂。残余物经硅胶柱层析(石油醚/乙酸乙酯=4:1)得到2.5g白色固体化合物a-1,收率为73%。熔点:93-94℃.1h nmr(400mhz,cdcl3)δ7.29(dd,j=8.4,2.4hz,1h),7.26
–
7.21(m,2h),7.15
–
7.08(m,2h),7.01
–
6.95(m,2h),4.93(t,j=7.8hz,1h),4.56
–
4.49(m,1h),4.40
–
4.32(m,1h),4.00(s,2h),2.75
–
2.67(m,1h),2.52
–
2.41(m,1h).
13
c nmr(101mhz,cdcl3)δ173.5,156.0,140.9,133.6,133.1,132.5,130.9,130.7,130.1,120.5,116.1,72.6,65.3,38.2,29.9.hrms(esi)calcd for c
17h19
brclo3na([m+na]
+
):402.9707;found:402.9710.
[0047]
实施例3:
[0048][0049]
在氩气保护下,向氢化反应内管中依次加入化合物a-1(1.14g,3.0mmol),手性螺环噁唑啉催化剂6c(3.0mg,3.0μmol)、叔丁醇钾(336mg,3.0mmol)、20ml正丙醇。密封反应釜,用氢气快速置换反应釜中的气体三次,调节氢气压力为10atm,室温下搅拌至氢气压力不再下降。反应结束后,缓慢释放出反应釜中的氢气,旋转蒸发仪脱除溶剂后得粗产物。柱层析得到932mg油状液体化合物c,收率80%,ee值93%。(c 0.5,etoh).1h nmr(400mhz,cdcl3)δ7.31
–
7.25(m,2h),7.23(d,j=8.4hz,1h),7.09(d,j=8.8hz,2h),6.91(d,j=8.4hz,2h),4.59
–
4.52(m,1h),3.99(s,2h),3.89
–
3.73(m,4h),2.38
–
1.89(m,4h).
13
c nmr(101mhz,cdcl3)δ156.5,141.1,133.6,133.1,131.5,130.9,130.6,130.1,120.5,116.3,76.4,64.2,59.0,38.2,33.8.hrms(esi)calcd for c
17h19
brclo3([m+h]
+
):385.0201;found:385.0199.hplc conditions:chiralcel ic-3column(25cm
×
0.46cmid);n-hexane/2-propanol=90:10;temp,rt;flow rate=1.0ml/min;210nmuv detector;tr(r)=12.0min(minor);tr(s)=13.7min(major).
[0050]
实施例4:
[0051]
[0052]
往10ml带支口的封管中依次加入化合物c(384mg,1.0mmol),无水对甲苯磺酸(86mg,0.5mmol)。氩气保护下加入干燥的甲苯(2ml),拧紧封管。于110℃下反应24h。冷却至室温,加入水,乙酸乙酯萃取三次(2ml
×
3)。无水硫酸镁干燥,抽滤,减压脱除溶剂得到粗品。粗品经硅胶柱层析分离提纯(石油醚/乙酸乙酯=5:1)得到329mg无色液体化合物d,产率90%。(c 0.5,chcl3).1h nmr(400mhz,cdcl3)δ7.31
–
7.20(m,3h),7.09(d,j=8.6hz,2h),6.80(d,j=8.8hz,2h),4.93
–
4.86(m,1h),4.02
–
3.94(m,5h),3.93
–
3.86(m,1h),2.25
–
2.10(m,2h).
13
c nmr(101mhz,cdcl3)δ156.1,141.2,133.6,133.1,130.9,130.6,130.1,120.5,115.5,77.3,73.2,67.2,38.2,33.0.hrms(esi)calcd for c
17h20
brclno2([m+nh4]
+
):384.0380;found:384.0368.
[0053]
实施例5:
[0054][0055]
手套箱中称取1.0mg手性催化剂和2.6mg配体,加入氢化釜内管中,用封口膜封口带出手套箱。迅速置于氢化釜内,封釜,加入3ml n
proh,经氮气置换三次后,体系于室温搅拌30min,再用氢气置换三次,充入20atm氢气,体系于室温下再搅拌30min,另取氢化内管,手套箱中称取1.19g式a化合物和336mg t
buok,用封口膜封口带出手套箱,迅速置于氢化釜内,封釜。氢化釜置换为氮气氛围,搅拌下向体系加入18ml n
proh,然后再加入3ml手性催化剂6c溶液,使用氢气置换三次,充入10atm氢气,体系于室温下搅拌12h后,氢气压力无明显下降。泄压开釜,将反应液于0℃下缓慢加入到半饱和氯化铵溶液中淬灭,使用dcm萃取三次,合并有机相使用无水硫酸钠干燥,过滤,滤液减压浓缩除尽溶剂。为式b化合物粗品。式b化合物粗品和0.72ml et3sih溶于21ml dcm,体系用冰浴降温,搅拌下逐滴加入0.56ml bf
3-et2o,加完体系于冰浴下继续搅拌3h。加水淬灭反应,两相分液,水相再用dcm萃取两次,合并有机相减压浓缩除尽溶剂。剩余物柱层析(石油醚/乙酸乙酯=3:1-0:1),得黄色油状物660mg。两步反应总收率57%,ee值89.7%。(c 0.5,etoh).1h nmr(400mhz,cdcl3)δ7.31
–
7.25(m,2h),7.23(d,j=8.4hz,1h),7.09(d,j=8.8hz,2h),6.91(d,j=8.4hz,2h),4.59
–
4.52(m,1h),3.99(s,2h),3.89
–
3.73(m,4h),2.38
–
1.89(m,4h).
13
c nmr(101mhz,cdcl3)δ156.5,141.1,133.6,133.1,131.5,130.9,130.6,130.1,120.5,116.3,76.4,64.2,59.0,38.2,33.8.hrms(esi)calcd for c
17h19
brclo3([m+h]
+
):385.0201;found:385.0199.hplc conditions:chiralcelic-3column(25cm
×
0.46cm id);n-hexane/2-propanol=90:10;temp,rt;flow rate=1.0ml/min;210nm uv detector;tr(r)=12.0min
(minor);tr(s)=13.7min(major).
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1