联苯基苯并咪唑并氮杂糖衍生物及其合成方法和应用
1.本发明涉及氮杂糖衍生物及其合成方法和应用,具体地说是涉及一种联苯基苯并咪唑并氮杂糖衍生物及其合成方法和应用。
背景技术:
2.糖类化合物广泛存在于各种细胞的表层,是细胞间传递信息的主要因子,在细胞信号转导、信息识别以及分裂、增殖、迁移等生理过程中发挥着重要作用。糖苷酶作为特异性糖苷键水解酶直接参与糖基化过程,许多重大疾病如糖尿病、肿瘤、病毒性感染、免疫性疾病等的发生、发展与糖苷酶的非正常表达密切相关(hudak,j.e.,et al.,chem.biol.,2013,21,16
‑
37.)。例如,酸性β
‑
葡萄糖苷酶、α
‑
半乳糖苷酶等的异常表达则会引起溶酶体贮积症(lysosomal storage disorder)。因此,以糖苷酶为靶点,设计合成其抑制剂,研究糖苷酶抑制活性及其抗肿瘤、抗糖尿病、抗病毒和免疫调节活性等,具有重要的意义和应用前景。
3.近年来,氮杂糖类衍生物通过抑制糖苷酶或糖基转移酶的活性,具有良好的生物学作用而成为人们研究的热点,如单环氮杂糖野尻霉素(1
‑
deoxynojirimycin,dnj)、双环氮杂糖苦马豆素(swainsonine,sw)、精氨栗碱(castanospermine,cas)(s.p.,et al.,j.am.chem.soc.,2018,140,5045
‑
5048.)以及多环(三元以上)稠杂氮杂糖如苯并咪唑并氮杂糖等(prasad,s.s.,et al.,j.org.chem.,2018,83,9604
‑
9618.)。目前,在单、双环氮杂糖结构改造以及构效关系研究方面已有大量工作,但稠杂氮杂糖的研究多仅限于合成方法学研究(yadav,l.d.s.,et al.,carbohydr.res.,2010,345,318
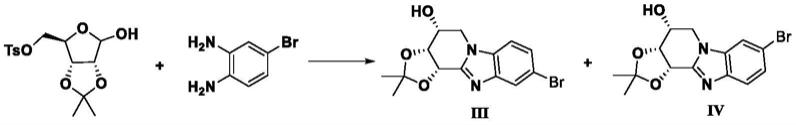
‑
323.),而对于其生物活性及结构改造则鲜有报道。作为我们在稠杂氮杂糖研究工作的延续(陈华等,zl201910014061.6),本发明采用新的合成途径,制备了以d
‑
核糖为糖原料的新型联苯基苯并咪唑并氮杂糖。化合物具有优越的β
‑
葡萄糖苷酶抑制活性,也是该类化合物作为糖苷酶抑制剂的首次研究,对寻求更多具有抗β
‑
葡萄糖苷酶活性的化合物,具有重要的意义。
技术实现要素:
4.本发明的目的是提供一种联苯基苯并咪唑并氮杂糖衍生物,其化学通式如式(i)或式(ii)所示:
[0005][0006]
其中,r为氢原子、甲基、甲氧基或羟基。
[0007]
上述的苯并咪唑并氮杂糖衍生物在制备β
‑
葡萄糖苷酶抑制剂类药物制剂中的应
用。
[0008]
试验表明上述化合物具有良好的β
‑
葡萄糖苷酶抑制活性,故可以在制备抗糖苷酶异常表达引起的相关疾病药物制剂中得以应用。
[0009]
本发明化合物与药理允许使用的载体均匀混合,按照常规的制剂方法可以制备成用于抗糖苷酶异常表达引起的相关疾病的各种形式的药物制剂。
[0010]
如本发明化合物为活性成份,可与水、蔗糖、山梨醇糖、果糖等组分组合制备成口服液体制剂;与赋形剂(乳糖、葡萄糖、蔗糖、甘露醇糖)、崩解剂(淀粉)、润滑剂(硬脂酸、滑石粉)、粘合剂(明胶、聚乙烯醇)等组分组合制备成片剂或胶囊剂。
[0011]
本发明化合物作为活性成份还可与生理盐水、葡萄糖溶液或盐水和葡萄糖组成的混合载体制备成注射液。
[0012]
本发明在用于临床时可参考的有效剂量是10~20mg/人/日,每日2~3次。医师也可依据患者个体差异,拟定服用剂量。
[0013]
一种联苯基苯并咪唑并氮杂糖衍生物的合成方法,该方法以丙叉基保护且对甲苯磺酰化(ts化)的d
‑
核糖与4
‑
溴
‑
1,2
‑
邻苯二胺为起始原料,经一步成环,得到化学通式如(iii)和(iv)所示的中间体化合物。化学通式如(iii)和(iv)所示的中间体化合物,分别再经与(含取代基)苯硼酸suzuki偶联、酸性条件脱除丙叉基,得到目标产物,其反应通式如下:
[0014][0015][0016]
所述合成方法具体包括以下步骤:
[0017]
(a)以丙叉基保护且对甲苯磺酰化的d
‑
核糖与4
‑
溴
‑
1,2
‑
邻苯二胺为起始原料,将原料在甲苯溶剂中溶解,加入三氟甲磺酸钪搅拌均匀,并于60
‑
100℃油浴下搅拌反应5
‑
10小时,加入饱和碳酸氢钠溶液中和,然后用二氯甲烷萃取,经减压蒸去溶剂,经柱色谱分离后得到化学通式如式(iii)和式(iv)所示的中间体化合物;
[0018][0019]
(b)将步骤(a)合成的化合物和苯硼酸按照摩尔比1∶1
‑
3,在水和1,4
‑
二氧六环中溶解,室温搅拌下,抽真空,充氮气,加入碳酸铯和催化剂pd(dppf)2cl2,然后在60
‑
100℃油浴下搅拌反应3
‑
6小时;加入乙酸乙酯和水萃取,有机相用无水硫酸钠干燥,抽滤并减压蒸去溶剂,经柱色谱分离后得到化学通式如式(v)和式(vi)所示的中间体化合物;
[0020][0021]
其中,r为氢原子、甲基或甲氧基;
[0022]
(c)将步骤(b)合成的化合物在酸性条件下脱除保护基,得到化学通式如式(i)和式(ii)所示的化合物;
[0023][0024]
其中,r为氢原子、甲基、甲氧基或羟基。
[0025]
步骤(a)中,所述d
‑
核糖与三氟甲磺酸钪的摩尔比为10∶1
‑
3。
[0026]
步骤(a)中,200
‑
300目硅胶柱色谱分离流动相为:v
石油醚
∶v
乙酸乙酯
=3
‑
1∶1。
[0027]
步骤(b)中,所述步骤(a)合成的化合物与碳酸铯的摩尔比为1∶3。
[0028]
步骤(b)中,所述步骤(a)合成的化合物与催化剂pd(dppf)2cl2的摩尔比为10∶1。
[0029]
步骤(b)中,200
‑
300目硅胶柱色谱分离流动相为:v
二氯甲烷
∶v
乙酸乙酯
=3
‑
2∶1。
[0030]
本发明的苯并咪唑并氮杂糖衍生物的制备方法高效、简便,化合物经柱色谱分离可得到该化合物异构体的纯品。该类化合物具有优越的β
‑
葡糖糖苷酶抑制活性,以及很强的抗糖苷酶异常表达引起的相关疾病的作用,以期为临床治疗糖苷酶异常表达引起的相关疾病提供更多的用药选择。
具体实施方式
[0031]
下面结合实施例对本发明做进一步的阐述,下述实施例仅作为说明,并不以任何方式限制本发明的保护范围。
[0032]
在下述实施例中未详细描述的过程和方法是本领域公知的常规方法,实施例中所用试剂均为分析纯或化学纯,且均可市购或通过本领域普通技术人员熟知的方法制备。下述实施例均实现了本发明的目的。
[0033]
实施例1
[0034]
合成(3ar,4r,11bs)
‑9‑
溴
‑
2,2
‑
二甲基
‑
3a,4,5,11b
‑
四氢苯并[4,5]咪唑[1,2
‑
a][1,3]二氧并[4,5
‑
c]吡啶
‑4‑
醇(简称化合物3)和(3ar,4r,11bs)
‑8‑
溴
‑
2,2
‑
二甲基
‑
3a,4,5,11b
‑
四氢苯并[4,5]咪唑[1,2
‑
a][1,3]二氧并[4,5
‑
c]吡啶
‑4‑
醇(简称化合物4)
[0035]
其化学反应流程如下:
[0036][0037]
具体方法为:
[0038]
称取丙叉基保护且对甲苯磺酰化(ts化)的d
‑
核糖1(4.3g,12.5mmol,市购商品或以d
‑
核糖为原料,参照文献方法aravind,a.,et al.,eur.j.org.chem.,2011,83,6980
‑
6988制备)和4
‑
溴
‑
1,2
‑
邻苯二胺(2.79g,1.2当量,市购商品)于250ml烧瓶中,加入80ml甲苯搅拌溶解,称取三氟甲磺酸钪(0.62g,0.1当量,市购商品),油浴升温至80℃,氮气保护。10h后,经tlc监测,原料d
‑
核糖反应完全。饱和碳酸氢钠溶液中和,二氯甲烷萃取反应液。减压蒸去溶剂,200目硅胶柱色谱分离(v
石油醚
∶v
乙酸乙酯
=1∶1),得到黄色油状化合物3和化合物4。
[0039]
化合物3:黄色油状物,收率23%。1h nmr(400mhz,cdcl3)δ(ppm):7.92(d,j=2.0hz,1h),7.42(dd,j=8.8,2.0hz,1h),7.28
‑
7.24(m,1h),5.47(d,j=6.8hz,1h),4.84(dd,j=6.8,2.8hz 1h),4.31
‑
4.27(m,1h),4.25
‑
4.20(m,1h),4.15
‑
4.12(m,1h),1.50(s,3h),1.36(s,3h);
13
c nmr(100mhz,cdcl3),δ(ppm):148.9,144.6,132.9,126.5,123.1,116.0,110.9,74.7,70.1,66.4,42.9,26.6,25.0;ms(esi):c
14
h
16
brn2o3([m+h]
+
):339.4.
[0040]
化合物4:黄色油状物,收率23%。1h nmr(400mhz,cdcl3)δ(ppm):7.62(d,j=8.8hz,1h),7.52(d,j=2.0hz,1h),7.38(dd,j=8.8,2.0hz,1h),5.47(d,j=6.8hz 1h),4.91
‑
4.85(m,1h),4.81(dd,j=7.2,3.2hz,1h),4.34
‑
4.31(m,1h),4.25
‑
4.21(m,1h),1.50(s,3h),1.38(s,3h);
13
c nmr(100mhz,cdcl3),δ(ppm):148.5,142.4,126.4,121.6,116.7,112.9,111.5,74.6,70.1,66.5,43.0,26.6,25.0;ms(esi):c
14
h
16
brn2o3([m+h]
+
):339.1.
[0041]
实施例2
[0042]
合成(3ar,4r,11bs)
‑9‑
(3,4
‑
二甲氧基苯基)
‑
2,2
‑
二甲基
‑
3a,4,5,11b
‑
四氢苯并[4,5]咪唑并[1,2
‑
a][1,3]二氧并[4,5
‑
c]吡啶
‑4‑
醇(简称化合物6a)
[0043]
其化学反应流程如下:
[0044][0045]
具体方法为:
[0046]
称取化合物3(0.3g,0.9mmol)和3,4
‑
二甲氧基苯硼酸(0.19g,1.2当量,市购商品)于25ml烧瓶中,加入水和1,4
‑
二氧六环共7ml做溶剂,加入碳酸铯(0.87g,3.0当量),抽真空,充氮气,后加入pd(ppf)cl2(0.07g,0.1当量,市购商品),抽真空,充氮气,升温至95℃反应,tlc监测原料化合物3反应完全,用水和乙酸乙酯萃取,取有机相,无水硫酸钠干燥,减压蒸去溶剂,300目硅胶柱色谱分离(v
二氯甲烷
∶v
乙酸乙酯
=2∶1),得到黄色固体产物6a。
[0047]
化合物6a:黄色固体,收率75%。1h nmr(400mhz,cdcl3)δ(ppm):7.78(d,j=8.4hz,1h),7.49
‑
7.46(m,2h),7.15
‑
7.11(m,2h),6.93(d,j=8.4hz,1h),5.48(d,j=6.8hz,1h),5.28(s,1h),4.83(dd,j=6.8,2.8hz,1h),4.35(dd,j=10.8,4.0hz,1h),4.25
‑
4.21(m,1h),3.95(s,3h),3.92(s,3h),1.46(s,3h),1.34(s,3h);
13
c nmr(100mhz,cdcl3),δ(ppm):149.3,148.7,142.5,137.1,134.6,134.4,122.8,120.2,119.8,111.6,111.3,110.9,107.7,70.3,66.5,56.1,53.5,42.8,26.7,25.1,25.1;ms(esi):c
22
h
24
n2o5([m+h]
+
):397.1.
[0048]
实施例3
[0049]
合成(2r,3r,4s)
‑7‑
(3,4
‑
二甲氧基苯基)
‑
1,2,3,4
‑
四氢苯并[4,5]咪唑并[1,2
‑
a]吡啶
‑
2,3,4
‑
三醇(简称化合物7a)
[0050]
其化学反应流程如下:
[0051][0052]
具体方法为:
[0053]
称取化合物6a(0.26g,0.66mmol)于25ml烧瓶中,加入30%三氟乙酸水溶液4ml溶解。氮气保护下室温反应,反应5h后,tlc监测原料反应完全。碳酸氢钠中和反应液,抽滤除去盐,滤液减压蒸去溶剂,200目硅胶柱色谱分离(v
二氯甲烷
∶v
甲醇
=15∶1),得到白色固体7a。
[0054]
化合物7a:白色固体,收率50%。(c 1mg/ml,ch3oh)。1h nmr(400mhz,dmso
‑
d6)δ(ppm):7.82
‑
7.75(m,1h),7.61(dd,j=8.4,2.0hz,1h),7.52
‑
7.47(m,1h),7.30
‑
7.21(m,2h),7.03(d,j=8.4hz,1h),4.82
‑
4.79(m,1h),4.31
‑
4.27(m,2h),4.24
‑
4.19(m,1h),3.88
‑
3.84(m,4h),3.79(s,3h);
13
c nmr(100mhz,dmso
‑
d6),δ(ppm):154.3,149.5,148.6,135.4,134.9,134.9,134.3,121.2,119.5,119.1,112.6,111.2,108.4,72.2,66.7,66.4,56.1,56.0,44.4,;ms(esi):c
19
h
20
n2o5([m+na]
+
):380.1.
[0055]
实施例4
[0056]
合成(2r,3r,4s)
‑7‑
(3,4
‑
二羟基苯基)
‑
1,2,3,4
‑
四氢苯并[4,5]咪唑并[1,2
‑
a]吡啶
‑
2,3,4
‑
三醇(简称化合物8a)。
[0057]
其化学反应流程如下:
[0058]
[0059]
具体方法为:
[0060]
称取6a(0.26g,0.66mmol)于25ml烧瓶中,加入5ml二氯甲烷溶解。抽真空,充氮气,冰浴搅拌下加入三溴化硼(0.07ml,3.0当量),10min后撤去冰浴,室温继续反应3h后,tlc监测原料反应完全。甲醇淬灭反应,碳酸氢钠固体中和反应液后抽滤,滤液减压蒸去溶剂,200目硅胶柱色谱分离(v
石油醚
∶v
甲醇
=5∶1),得到白色固体8a。
[0061]
化合物8a:白色固体,收率40%。(c 1mg/ml,ch3oh)。1h nmr(400mhz,dmso
‑
d6)δ(ppm):8.03(s,1h),7.74(s,2h),7.17(d,j=2.4hz,1h),7.07(dd,j=8.4,2.4hz,1h),6.86(d,j=8.4hz,1h),5.20(s,1h),4.54(q,j=6.0hz,1h),4.36
‑
4.32(m,1h),4.16(s,1h),3.95(t,j=10.8hz,1h);
13
c nmr(100mhz,dmso
‑
d6),δ(ppm):153.2,146.2,146.1,138.9,132.5,131.1,130.8,125.2,118.9,116.6,115.0,115.0,110.2,71.2,66.2,65.3,44.6;ms(esi):c
17
h
16
n2o5([m+k]
+
):367.1.
[0062]
实施例5
[0063]
合成(2r,3r,4s)
‑8‑
(3,4
‑
二甲氧基苯基)
‑
1,2,3,4
‑
四氢苯并[4,5]咪唑并[1,2
‑
a]吡啶
‑
2,3,4
‑
三醇(简称化合物7b)。
[0064]
其化学反应流程如下:
[0065][0066]
具体方法为:
[0067]
称取化合物4(实施例1合成的化合物4)和3,4
‑
二甲氧基
‑
苯硼酸5a,按实施例2的方法,合成化合物6b。再称取化合物6b,按实施例3的方法,合成化合物7b。
[0068]
化合物7b:白色固体,收率48%。1h nmr(400mhz,dmso
‑
d6)δ(ppm):7.83
‑
7.81(m,1h),7.54
‑
7.50(m,2h),7.24
–
7.19(m,2h),7.02(d,j=8.4hz,1h),5.79
‑
8.71(m,1h),5.54
‑
5.51(m,1h),5.35
‑
5.32(m,1h),4.82(s,1h),4.24
‑
4.20(m,2h),4.12(s,1h),3.88
‑
3.83(m,4h),3.78(s,3h);
13
c nmr(100mhz,dmso
‑
d6),δ(ppm):154.2,149.3,148.2,143.9,134.6,134.3,133.8,121.2,119.2,116.4,112.5,110.9,110.5,71.9,66.5,66.1,55.8,55.8,48.9,44.1;ms(esi):c
19
h
20
n2o5([m+h]
+
):357.3.
[0069]
实施例6
[0070]
合成(2r,3r,4s)
‑8‑
(3,4
‑
二羟基苯基)
‑
1,2,3,4
‑
四氢苯并[4,5]咪唑并[1,2
‑
a]吡啶
‑
2,3,4
‑
三醇(简称化合物8b)。
[0071]
其化学反应流程如下:
[0072]
[0073]
具体方法为:
[0074]
称取化合物6b,按实施例3的方法,合成化合物8b。
[0075]
化合物8b:白色固体,收率39%。(c 1mg/ml,ch3oh)。1h nmr(400mhz,dmso
‑
d6)δ(ppm):7.97(d,j=8.4hz,1h),7.79
‑
7.73(m,2h),7.11(d,j=2.4hz,1h),7.01(dd,j=8.4,2.4hz,1h),6.87(d,j=8.0hz,1h),5.24(s,1h),4.52(q,j=5.6hz,1h),4.35(t,j=8.8hz,1h),4.17
‑
4.16(m,1h),3.95(t,j=10.8hz,1h);
13
c nmr(100mhz,dmso
‑
d6),δ(ppm):153.2,146.3,146.3,139.9,132.0,130.8,130.3,125.0,118.8,116.7,114.9,113.9,110.8,71.1,66.2,65.2,44.6;ms(esi):c
17
h
16
n2o5([m+k]
+
):367.1.
[0076]
实施例7
[0077]
合成(2r,3r,4s)
‑7‑
苯基
‑
1,2,3,4
‑
四氢苯并[4,5]咪唑并[1,2
‑
a]吡啶
‑
2,3,4
‑
三醇(简称化合物7c)
[0078]
化学反应流程如下:
[0079][0080]
具体方法为:
[0081]
称取化合物3(实施例1合成的化合物3)和苯硼酸5b,按实施例2的方法,合成化合物6c。再称取化合物6c,按实施例3的方法,合成化合物7c。
[0082]
化合物7c:白色固体,收率52%。m.p.200.0
‑
201.1℃,(c 1mg/ml,ch3oh),1h nmr(600mhz,dmso
‑
d6)δ(ppm):7.85(s,1h),7.68(d,j=7.8hz,2h),7.59
‑
7.52(m,2h),7.50
‑
7.45(m,2h),7.33(t,j=7.2hz,1h),4.83(d,j=3.0hz,1h),4.25
‑
4.21(m,2h),4.14(d,j=3.6hz,1h),3.87(t,j=12.0hz,1h);
13
c nmr(150mhz,dmso
‑
d6),δ(ppm):154.2,143.7,141.2,134.4,134.0,128.9,126.9,126.7,121.8,121.2,119.6,116.6,110.4,71.5,66.3,66.0,44.0;ms(esi):c
17
h
16
n2o3([m+h]
+
):297.2.
[0083]
实施例8
[0084]
合成(2r,3r,4s)
‑8‑
苯基
‑
1,2,3,4
‑
四氢苯并[4,5]咪唑并[1,2
‑
a]吡啶
‑
2,3,4
‑
三醇(简称化合物7d)
[0085]
化学反应流程如下:
[0086][0087]
具体方法为:
[0088]
称取化合物4(实施例1合成的化合物4)和苯硼酸5b,按实施例2的方法,合成化合
物6d。再称取化合物6d,按实施例3的方法,合成化合物7d。
[0089]
化合物7d:白色固体,收率48%。m.p.144.1
‑
145.2℃,(c 1mg/ml,ch3oh),1h nmr(600mhz,dmso
‑
d6)δ(ppm):8.45(s,2h),7.78
‑
7.73(m,2h),7.65(d,1h),7.51
‑
7.45(m,2h),7.34(t,j=7.2hz,1h),4.82(d,j=3.0hz,1h),4.30
‑
4.27(m,1h),4.24
‑
4.20(m,1h),4.13(d,j=3.0hz,1h)3.89(t,j=9.6hz,1h);
13
c nmr(150mhz,dmso
‑
d6),δ(ppm):154.2,142.7,141.1,135.0,134.5,128.9,127.0,126.9,121.8,121.0,118.9,110.1,108.3,71.6,66.3,66.0,44.1;ms(esi):c
17
h
16
n2o3([m+h]
+
):297.0
[0090]
实施例9
[0091]
合成(2r,3r,4s)
‑7‑
(邻甲苯基)
‑
1,2,3,4
‑
四氢苯并[4,5]咪唑并[1,2
‑
a]吡啶
‑
2,3,4
‑
三醇(简称化合物7e)
[0092]
其化学反应流程如下:
[0093][0094]
具体方法为:
[0095]
称取化合物3(实施例1合成的化合物3)和2
‑
甲基
‑
苯硼酸5c,按实施例2的方法,合成化合物6e。再称取化合物6e,按实施例3的方法,合成化合物7e。
[0096]
化合物7e:白色固体,收率47%。m.p.148.4
‑
149.9℃,(c 1mg/ml,ch3oh),1h nmr(600mhz,dmso
‑
d6)δ(ppm):7.52(t,j=8.4hz,1h),7.28
‑
7.23(m,4h),7.17(d,j=8.4hz,2h),4.83(s,1h),4.25
‑
4.22(m,2h),4.14(s,1h),3.88(t,j=12.6hz,1h),2.22(s,3h);
13
c nmr(150mhz,dmso
‑
d6),δ(ppm):154.0,143.1,142.2,135.2,135.0,133.4,130.3,130.0,127.0,125.9,123.3,118.9,109.8,71.6,66.3,66.0,44.0,20.4;ms(esi):c
18
h
18
n2o3([m+h]
+
):311.1.
[0097]
实施例10
[0098]
合成(2r,3r,4s)
‑8‑
(邻甲苯基)
‑
1,2,3,4
‑
四氢苯并[4,5]咪唑并[1,2
‑
a]吡啶
‑
2,3,4
‑
三醇(简称化合物7f)。
[0099]
其化学反应流程如下:
[0100][0101]
具体方法为:
[0102]
称取化合物4(实施例1合成的化合物4)和2
‑
甲基
‑
苯硼酸5c,按实施例2的方法,合成化合物6f。再称取化合物6f,按实施例3的方法,合成化合物7f。
[0103]
化合物7f:白色固体,收率53%。m.p.79.9
‑
81.1℃,(c 1mg/ml,ch3oh),1h nmr(600mhz,dmso
‑
d6)δ(ppm):7.62(d,j=13.8hz,1h),7.42(s,1h),7.30
‑
7.25(m,4h),7.14(d,j=7.8hz,1h),4.82(d,j=3.0hz,1h),4.23
‑
4.20(m,2h),4.13(s,1h),3.85(t,j=12.0hz,1h),2.25(s,3h);
13
c nmr(150mhz,dmso
‑
d6),δ(ppm):153.9,142.0,142.0,135.2,134.9,134.3,130.2,129.9,127.0,125.8,123.0,118.0,110.4,71.5,66.3,66.0,62.8,44.0;ms(esi):c
18
h
18
n2o3([m+h]
+
):311.0.
[0104]
实施例11
[0105]
本发明化合物对β
‑
葡糖糖糖苷酶抑制活性测试
[0106]
测试方法:
[0107]
β
‑
葡萄糖苷酶(杏仁)及其对硝基苯酚
‑
β
‑
葡萄糖苷底物(pnpg)均购为sigma公司产品。实验分为空白组、对照组和样品组,每组3个平行。在96孔板中依次加入ph=7.3磷酸盐缓冲液(pbs)缓冲液、抑制剂溶液和酶溶液,共50μl混匀,于37℃保温10min。再加入底物溶液50μl,混匀后于37℃恒温20min。结束后加入100μl 1mol/l的na2co3溶液中止反应。由于pnpg在β
‑
葡萄糖苷酶的作用下能水解产生葡萄糖和对硝基酚(pnp),通过酶标仪在od
405nm
下测定样品的吸光率。样品糖苷酶抑制活性%=100
‑
[(样品组od 405nm
/空白组od 405nm
)
×
100]。
[0108]
测试结果:见表1。
[0109]
表1本发明化合物(7a~7f)和(8a~8b)对β
‑
葡萄糖苷酶抑制活性
[0110][0111]
注释a:化合物在100μm浓度时的抑制率。
[0112]
由表1可见,本发明化合物对β
‑
葡萄糖苷酶均具有较强的抑制活性,尤其是化合物7b,显示出更强的抑制活性,对β
‑
葡萄糖苷酶抑制的ic
50
值达到0.09μm,是临床药物米格列醇(miglitol)的活性的10倍左右,提示这个化合物可能具有更好的抗糖苷酶异常表达引起的相关疾病活性。这是联苯基苯并咪唑并氮杂糖化合物作为糖苷酶抑制剂的首次报道,对于后续高活性的β
‑
葡萄糖苷酶抑制剂的设计、合成具有重要的意义。
[0113]
实施例12
[0114]
取实施例5制备的化合物7b 5mg,乳糖60mg,马铃薯粉30mg,聚乙烯醇2mg,硬脂酸镁1mg,制备成口服片剂。
[0115]
本发明列举的实施例1
‑
12旨在阐明苯并咪唑并氮杂糖衍生物的制备方法以及该类化合物对β
‑
葡萄糖苷酶的抑制活性,实施例不单是说明它本身所述的具体的化合物的合成方法及抗β
‑
葡萄糖苷酶活性,同时也可以用来说明改变原料的种类和数量,合成其同系
物和类似物,而不对本发明的范围构成任何限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1