一种有机共轭聚合物及其制备方法和应用
本发明涉及光催化水制氢技术领域,更具体地,涉及一种有机共轭微孔聚合物及其制备方法和应用。
背景技术:
近年来,随着石油、煤炭、天然气等不可再生资源的不断消耗,以及化石燃料燃烧带来的环境污染问题,让人们意识到开发新型的清洁可再生能源至关重要。结合太阳能取之不尽、用之不竭的特点以及氢能源清洁无污染、燃烧值大、可储存运输的优点,开发光催化水制氢的催化剂逐渐进入人们的视野。目前用于光解水制氢的催化剂主要有无机半导体材料,有机共轭聚合物等。无机半导体材料光谱响应范围小、量子效率低等特点限制了它的发展,而有机共轭聚合物具有结构多样,易于修饰,传输电子性能高等特点,成为近年来研究的热点。构建给体-受体(d-a)型共轭聚合物是开发高效光催化剂的有效策略。d-a型有机共轭微孔聚合物具有高比表面积、合成方法多样、共轭程度高、电子传输能力强、可控的分子能隙等特点,在光催化水制氢领域具有重要的作用。
但是目前的有机共轭聚合物作为光催化剂制氢的速率并不高,限制了其大规模应用。例如中国专利(cn111804338a)公开了一种三嗪基d-a型含氮有机共轭多孔聚合物光催化材料及其制备与应用,主要采用的是三嗪基和吡唑小分子结合,从数据上看,其最高产氢速率(her)只有1000μmolh-1g-1。
技术实现要素:
本发明为克服光催化水制氢速率差的缺陷,提供一种有机共轭聚合物。
本发明的另一目的在于提供所述有机共轭聚合物的制备方法。
本发明的另一目的是提供一种包含有机共轭聚合物的光催化剂及其应用。
为实现上述目的,本发明采用的技术方案是:
一种有机共轭聚合物,所述有机共轭聚合物的结构式如式ⅰ所示,
其中x为o,s原子;n为50~10000;
其中
本发明所述有机共轭聚合物是由苯并双噻吩或者苯并双呋喃作为给体,二苯并[b,d]噻吩5,5-二氧化物作为受体,通过suzuki偶联反应所得。苯并双噻吩和苯并双呋喃芳香性好,单体结构稳定,通过
优选地,所述x为s原子。
s原子的电负性小于o原子,噻吩的芳香性比呋喃高,所以苯并双噻吩的稳定性比苯并双呋喃高,能够提高光解水制氢。
优选地,所述
优选地,所述有机共轭聚合物的孔径在2~60nm。
所述有机共轭聚合物的孔径在2~60nm时具有大的表面积,形状规整,能最大限度使聚合物在光催化水产氢过程中保持良好的稳定性,提高聚合物的产氢速率。
本发明还提供所述有机共轭聚合物的制备方法,包括如下步骤:
惰性气氛下,苯并双噻吩或者苯并双呋喃在钯催化剂中加入
优选地,所述苯并双噻吩或者苯并双呋喃与钯催化剂的摩尔比为1:(0.01~0.015)。
优选地,所述苯并双噻吩或苯并双呋喃与无机碱的摩尔比为1:(5~10)。
优选地,所述加热反应的反应温度为100~160℃,反应时间为12~60h。
所述无机碱与有机溶剂用量比例为1mmol:(4~6)ml。
一种光催化剂,包含所述有机共轭聚合物。
所述光催化剂在光催化分解水制氢中的应用。
与现有技术相比,本发明的有益效果是:
本发明提供了一种有机共轭聚合物,采用苯并双噻吩或者苯并双呋喃为母体和
附图说明
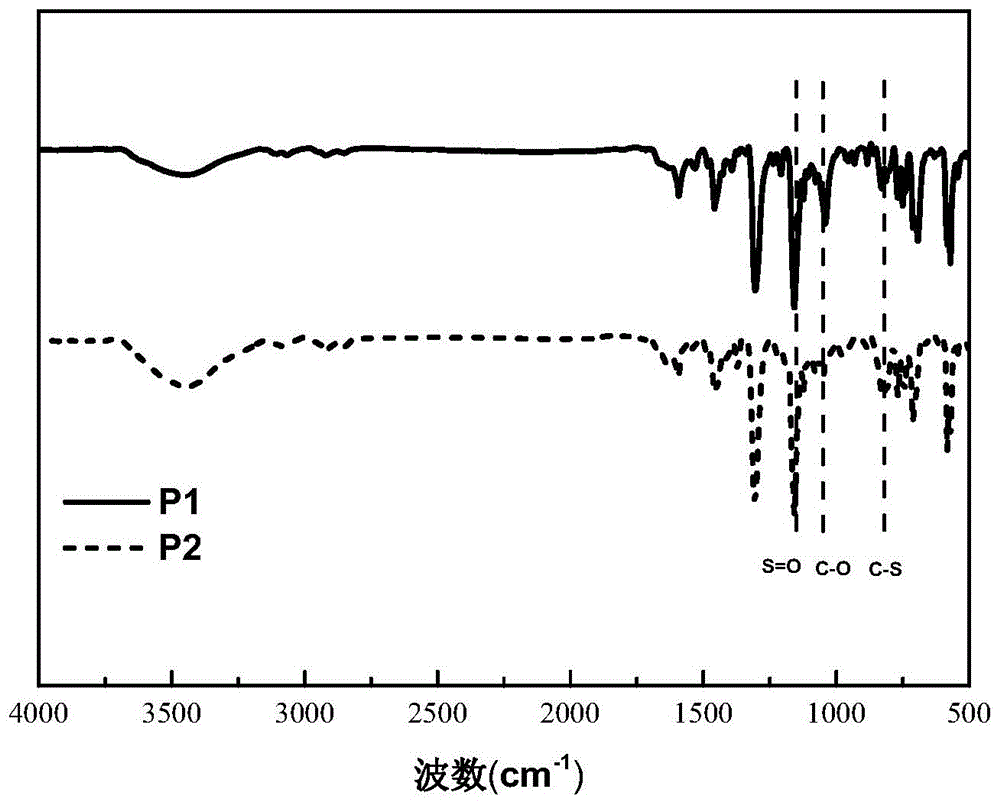
图1为实施例1~2的光催化剂p1、p2的红外光谱图;
图2为实施例1~2的光催化剂p1、p2的紫外-可见吸收(uv-vis)光谱图;
图3为实施例1~2的光催化剂p1、p2的光催化水分解产氢效率图。
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,以下实施例所用试剂和材料均为市购。
实施例1
聚合物p1的合成
合成路线如下:
准确称取单体m1(1.000mmol,0.3741g)和单体m2(0.500mmol,0.4131g),加入48ml厚壁耐压瓶中,在惰性气体环境下加入pd(pph3)4(0.025mmol,29mg),k2co3(2mol/l,2.5ml)以及dmf(15ml),密封。在150℃的条件下避光反应48h。反应液冷却至室温后,滴入不断搅拌的甲醇溶液中,过滤得到粗产物。粗产物依次用100ml甲醇、石油醚进行索氏抽提各24h,剩余物固体用甲醇冲洗,真空干燥24h,得到褐色粉末状产物p1,所述p1的聚合度为4。
本实施例的光催化剂由共聚物p1组成。
实施例2
聚合物p2的合成
代表性合成路线如下:
准确称取单体m1(1.000mmol,0.3741g)和单体m3(0.500mmol,0.4292g),加入48ml厚壁耐压瓶中,在惰性气体环境下加入pd(pph3)4(0.025mmol,29mg),k2co3(2mol/l,2.5ml)以及dmf(15ml),密封。在150℃的条件下避光反应48h。反应液冷却至室温后,滴入不断搅拌的甲醇溶液中,过滤得到粗产物。粗产物依次用100ml甲醇、石油醚进行索氏抽提各24h,剩余物固体用甲醇冲洗,真空干燥24h,得到黄色粉末状产物p2,所述p2的聚合度为4。
本实施例的光催化剂由共聚物p2组成。
实施例3
制备方法同实施例2,其区别在于,m1的单体替换成a2单体
本实施例的光催化剂由共聚物p3组成。
实施例4
制备方法同实施例2,其区别在于,m1的单体替换成a3单体
本实施例的光催化剂由共聚物p4组成。
实施例5
制备方法同实施例2,其区别在于,m1的单体替换成a4单体
本实施例的光催化剂由共聚物p5组成。
实施例6
制备方法同实施例2,其区别在于,m1的单体替换成a5单体
本实施例的光催化剂由共聚物p6组成。
实施例7
制备方法同实施例2,其区别在于,m1的单体替换成a6单体
本实施例的光催化剂由共聚物p7组成。
实施例8
制备方法同实施例2,其区别在于,m1的单体替换成a3单体
本实施例的光催化剂由共聚物p8组成。
实施例9
制备方法同实施例2,其区别在于,m1的单体替换成a3单体
本实施例的光催化剂由共聚物p9组成。
实施例10
制备方法同实施例2,其区别在于,m1的单体替换成a3单体
本实施例的光催化剂由共聚物p10组成。
对比例1
制备方法同实施例2,其区别在于,m3单体替换成
本对比例的光催化剂由共聚物p8组成。
对比例2
制备方法同实施例2,其区别在于,m1单体替换成
本对比例的光催化剂由共聚物p9组成。
上述实施例和对比例均经过性能测试
产氢速率的测试步骤:
称取5mg聚合物,加入50ml抗坏血酸溶液(0.2mol/l),超声震荡15分钟至聚合物完全分散,使用泊菲莱labsolar6a全玻璃自动在线微量气体分析系统测试,在全光谱照射下6小时,得到产氢速率结果。
表1实施例和对比例的数据
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!