一种手性N-Boc-吡咯烷-3-硼酸类化合物的制备方法与流程
一种手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸类化合物的制备方法
技术领域
1.本发明涉及制药技术领域,尤其涉及一种手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸类化合物的制备方法。
背景技术:
2.烷基硼酸衍生物是一类重要的中间体,在现代有机合成、医药和材料科学等领域有着广泛的应用。特别是烷基硼酸酯是一类非常多元化的合成砌块,可以通过偶联反应、氧化反应等衍生成c
‑
卤键、c
‑
杂键和c
‑
c键。经典的烷基硼酸酯的制备方法主要集中在非环状的烷基硼酸酯,主要包括烷基金属试剂的转金属化和亲电硼化(organometallics 1983,2,1311)、烯烃的硼氢化(chem.rev.1991,91,1179)、过渡金属催化的c(sp3)
–
h键活化硼化(j.organomet.chem.2003,680,3)、烷基卤化物的miyaura型硼化(angew.chem.int.ed.2012,51,528),以及2017年baran和aggarwal小组接连在science杂志上发表的脱羧硼化反应(science 2017,356,1045;science 2017,357,283)。然而,这些合成方法都有各自的缺点,如官能团兼容性不好、原料不容易获得、反应收率不高、难以实现工业放大生产、区域选择性或者对映选择性不好等问题。其中,特别是对于环状的烷基硼酸类化合物,其手性合成更具有挑战性,因此这也阻碍了环烷基硼酸类化合物的应用。
3.此外,在医药领域,硼酸作为羧酸的电子等排体,近年来越来越多地开始应用于药物的设计和开发,许多含有手性烷基硼酸结构的化合物的许多独特的生物活性已被揭示。其中,特别是手性α
‑
氨基硼酸类化合物受到了非常广泛的关注,它们被证明是蛋白酶体抑制剂的关键药效团,如用于治疗多发性骨髓瘤的药物硼替佐米(velcade,2003年上市)和伊莎佐米(ninlaro,2015年上市)。此外,据报道许多α
‑
氨基硼酸类化合物还具有良好的抗癌、抗病毒和抗菌活性。这些化合物的成功应用,进一步激发了药物化学家们对寻找含手性α
‑
氨基硼酸的生物活性小分子的越来越大的兴趣。从近十余年的文献和专利中也可以看出,关于含有手性α
‑
氨基硼酸的化合物的发现和在医药中的应用的报道数量呈逐年递增的趋势。
4.可以预见,手性β
‑
氨基硼酸类化合物与手性α
‑
氨基硼酸类化合物类似,很可能也具有类似的生物活性,具有非常大的应用潜力。然而,对于β
‑
氨基硼酸的合成方法(特别是手性合成方法)的文献和专利报道较少,这极大地限制了这类化合物的应用。目前,尚无含有手性β
‑
氨基硼酸结构的药物进入临床研究阶段或者上市。加拿大多伦多大学的yudin教授发表了一种β
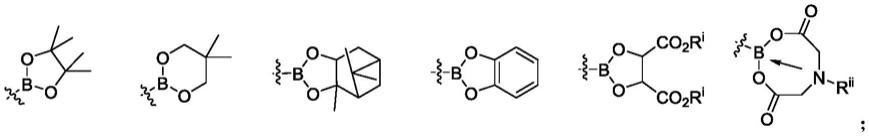
‑
氨基硼酸片段的合成方法学及其应用(angew.chem.int.ed.2016,55,12659
–
12663)。他们通过α
‑
醛基硼酸酯与含有裸露氨基的多肽化合物的还原胺化反应得到一系列含有β
‑
氨基硼酸片段的含硼多肽化合物,并发现这类化合物具有较好的稳定性。这种β
‑
氨基硼酸衍生物的温和的合成方法在蛋白酶体的可逆共价抑制剂的药物设计方面的应用具有较大的潜力,为新药设计人员开启了一扇新的窗户。不过,该方法需要用到制备工艺复杂(需要用到臭氧氧化)的α
‑
醛基硼酸酯原料,只能合成非环状的β
‑
氨基硼酸衍生物,且无法实现手性合成。因此,亟需开发更加高效、选择性更好、容易放大的β
‑
氨基硼酸的新
的合成方法,为这类化合物能早日应用于医药领域奠定基础。
技术实现要素:
5.有鉴于此,本发明要解决的技术问题在于提供一种手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸类化合物的制备方法,制备得到的手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸类化合物光学纯度高、收率高。
6.为达到上述目的,本发明提供了一种手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸类化合物的制备方法,包括:
7.s1)采用手性试剂(+)
‑
ipc2bh或(
‑
)
‑
ipc2bh,与式i所示n
‑
boc
‑3‑
吡咯啉进行不对称硼氢化反应,得到式ⅱ所示三烷基硼中间体;
8.s2)采用碱性水溶液与式ⅱ所示三烷基硼中间体反应,得到式ⅲ所示手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸;
[0009][0010]
上述反应的方程式如下:
[0011][0012]
所述步骤s1)具体为:采用手性试剂(+)
‑
ipc2bh或(
‑
)
‑
ipc2bh,与式i所示n
‑
boc
‑3‑
吡咯啉,在醚类溶剂(s1)中进行不对称硼氢化反应,得到式ⅱ所示三烷基硼中间体。
[0013]
所述醚类溶剂(s1)优选为四氢呋喃、乙醚和甲基叔丁基醚中的一种或多种。
[0014]
本发明中,所述手性试剂(+)
‑
ipc2bh或(
‑
)
‑
ipc2bh指二异松蒎烯基硼烷。
[0015]
本发明对上述手性试剂(+)
‑
ipc2bh或(
‑
)
‑
ipc2bh的来源并无特殊限定,可以为一般市售或按照本领域技术人员熟知的方法自行制备。
[0016]
其制备方法的反应方程式优选如下:
[0017][0018]
其中,所得的(+)
‑
ipc2bh或(
‑
)
‑
ipc2bh浓度优选为0.5m~10.0m。
[0019]
所述(+)
‑
α
‑
蒎烯或(
‑
)
‑
α
‑
蒎烯与bh3·
sme2的摩尔比优选为(2~3):1。
[0020]
所述反应温度(t4)优选为
‑
10~10℃,所述时间(t4)优选为3h~1天。
[0021]
值得注意的是,(+)
‑
ipc2bh或(
‑
)
‑
ipc2bh试剂制备完成后,需先在减压下除去大部分反应液中的四氢呋喃和释放的游离二甲硫醚,再将剩余物溶于醚类溶剂(s1)中参与下一步不对称硼氢化反应。实验证明,二甲硫醚的大量存在会大大降低终产物的光学纯度。可能的原因是,反应体系中存在ipc2bh和ipcbh2的平衡,而二甲硫醚与形成的ipc2bh配位后会抑制产生高光学纯度的ipc2bh的平衡(j.org.chem.1982,47,5065)。
[0022]
本发明优选的,所述手性试剂(+)
‑
ipc2bh或(
‑
)
‑
ipc2bh,与式i所示n
‑
boc
‑3‑
吡咯啉的摩尔比为(1~3):1。
[0023]
所述不对称硼氢化反应的温度(t1)优选为
‑
40~10℃;所述不对称硼氢化反应的时间(t1)优选为20h~3天。
[0024]
本发明优选的,不对称硼氢化反应后对体系进行纯化,具体的:
[0025]
减压下除去反应溶剂,得到油状粗品,即式ⅱ所示三烷基硼中间体。
[0026]
然后将得到的式ⅱ所示三烷基硼中间体溶解于醇类溶剂(s2)中,采用碱性水溶液与式ⅱ所示三烷基硼中间体反应。
[0027]
所述醇类溶剂(s2)优选为甲醇或乙醇。
[0028]
所述碱性水溶液优选为碳酸钾、碳酸氢钾、碳酸钠或碳酸氢钠的水溶液。
[0029]
所述碱性水溶液的浓度优选为0.05g/l~1.0g/l。
[0030]
本发明优选的,上述反应的温度(t2)优选为10~40℃;上述反应的时间(t2)优选为10min~1h。
[0031]
上述步骤s2)优选具体为:
[0032]
将步骤s1)得到的式ⅱ所示三烷基硼中间体用醇类溶剂和碱性水溶液的混合溶液溶解,进行反应。
[0033]
本发明优选的,反应后,进行萃取、浓缩,得到式ⅲ所示手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸。
[0034]
本发明优选的,所述萃取的溶剂选自石油醚或正己烷。
[0035]
本发明优选的,萃取后的有机相经醇类溶剂和碱性水溶液的混合溶液洗涤、分液,合并所得水相,并用石油醚或正己烷进行萃取、分液,以进一步除去杂质。
[0036]
此时目标中间体(ⅲ)在水相中以硼酸钠盐的形式存在,用稀盐酸溶液调节ph值至1~3后,加入固体氯化钠至饱和,再用乙酸乙酯多次萃取,有机相合并后用饱和食盐水洗涤、用无水硫酸钠干燥,过滤和浓缩后即可得到中间体(ⅲ)的油状粗品。必要情况下,可重复上述操作1~3次以提高目标中间体粗品的萃取回收率。
[0037]
本发明还提供了一种手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸类化合物的制备方法,包括:
[0038]
s1)采用手性试剂(+)
‑
ipc2bh或(
‑
)
‑
ipc2bh,与式i所示n
‑
boc
‑3‑
吡咯啉进行不对称硼氢化反应,得到式ⅱ所示三烷基硼中间体;
[0039]
s2)采用碱性水溶液与式ⅱ所示三烷基硼中间体反应,得到式ⅲ所示手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸;
[0040]
s3)将式ⅲ所示手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸与醇类化合物进行酯化反应,得到式
ⅳ‑
a或式
ⅳ‑
b所示手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸酯;
[0041][0042]
其中,r3、r4独立的优选为c1~c10的烷基;更优选为甲基、乙基、异丙基、叔丁基或苄基;
[0043]
r1、r2与硼原子和氧原子共同形成5~8元杂环;
[0044]
优选形成以下任一结构:
[0045][0046]
其中,r
i
、r
ii
独立的优选为氢原子、甲基、乙基、异丙基、叔丁基或苄基。
[0047]
上述反应的方程式如下:
[0048][0049]
上述步骤s1)、s2)同上,在此不再赘述。
[0050]
本发明中,所述醇类化合物可以选自单醇或二醇。
[0051]
具体的,所述醇类化合物优选为甲醇、乙醇、异丙醇、叔丁醇或苄醇,或者以下任一结构:
[0052][0053]
其中,r
i
、r
ii
独立的优选为氢原子、甲基、乙基、异丙基、叔丁基或苄基。
[0054]
得到的手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸酯具有以下式
ⅳ‑
a或式
ⅳ‑
b所示结构:
[0055][0056]
其中,r1、r2与硼和氧原子共同形成5~8元杂环。
[0057]
优选的,所述5~8元杂环选自以下任一结构:
[0058][0059]
其中,r
i
、r
ii
独立的优选为氢原子、甲基、乙基、异丙基、叔丁基或苄基。
[0060][0061]
其中,r3、r4优选为c1~c10的烷基;更优选独立的选自甲基、乙基、异丙基、叔丁基或苄基。
[0062]
本发明中,所述醇类化合物和式i所示n
‑
boc
‑3‑
吡咯啉的摩尔比优选为(0.8~
10):1。
[0063]
所述酯化反应的溶剂(s3)优选为乙酸乙酯、乙酸异丙酯或乙酸叔丁酯。
[0064]
所述酯化反应的温度(t3)优选为10~40℃;所述酯化反应的时间(t3)优选为0.5~2h。
[0065]
本发明优选的,酯化反应后进行纯化,具体的:
[0066]
将酯化反应得到的粗品经过减压浓缩和硅胶柱层析纯化得到手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸酯纯品。
[0067]
本发明优选的,所述柱层析为快速柱层析。
[0068]
所述柱层析的洗脱液优选为石油醚、乙酸乙酯混合溶剂。
[0069]
本发明中,与硼原子直接相连的碳原子(*标记)为手性碳,其构型为r或s。
[0070]
本发明提供的制备方法整个制备过程共4步,只需要对最后一步的产品进行一次柱层析纯化;而且,由于步骤s2)粗品经过碱性水溶液处理后,大部分杂质被除去,所得的粗品纯度极高,进一步降低了终产品的纯化难度;终产品只需经过较短的快速柱层析纯化,即可获得高纯度的目标化合物。
[0071]
本发明制备的手性化合物含有可进一步修饰的氨基和硼酸基团,作为一种重要的双官能团化的有机合成中间体,既可用于衍生化制备含有手性吡咯烷
‑3‑
硼酸片段的药物或者药物中间体;也可通过偶联反应转化为含有3
‑
位被烷基、烯基或者芳基等取代的手性吡咯烷的合成砌块,在有机合成、材料科学或者医药领域中都具有非常重要的应用价值。
[0072]
与现有技术相比,本发明提供了一种手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸类化合物的制备方法,包括:s1)采用手性试剂(+)
‑
ipc2bh或(
‑
)
‑
ipc2bh,与式i所示n
‑
boc
‑3‑
吡咯啉进行不对称硼氢化反应,得到式ⅱ所示三烷基硼中间体;s2)采用碱性水溶液与式ⅱ所示三烷基硼中间体反应,得到式ⅲ所示手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸;s3)将式ⅲ所示手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸与醇类化合物进行酯化反应,得到式
ⅳ‑
a或式
ⅳ‑
b所示手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸酯。
[0073]
本发明首次公开了一种n
‑
boc
‑
吡咯烷
‑3‑
硼酸类化合物的手性制备方法,所述制备方法具有以下优势:
[0074]
1)起始原料和试剂便宜易得;
[0075]
2)操作方法简单,重复性好;
[0076]
3)产物的光学纯度高,ee值>96%;
[0077]
4)可实现公斤级放大;
[0078]
5)纯化方法简便,4步反应只需最终一次柱层析,总收率高达60%以上。
具体实施方式
[0079]
为了进一步说明本发明,下面结合实施例对本发明提供的手性n
‑
boc
‑
吡咯烷
‑3‑
硼酸类化合物的制备方法进行详细描述。
[0080]
本发明所使用的物料缩写含义如下表:
[0081]
表1各物料缩写含义汇总
[0082][0083][0084]
本发明的制备方法中,各种优选条件可在不违背本领域常识的基础上任意组合,即可得本发明的较佳的实例。除特殊说明外,本发明所设计的原料和试剂均为市售。
[0085]
实施例1
[0086]
反应方程式如下:
[0087][0088]
步骤一:制备(
‑
)
‑
ipc2bh试剂
[0089]
‑
5℃,氮气保护下,将(+)
‑
α
‑
蒎烯(705ml)于45min内缓慢滴加到硼烷二甲硫醚络合的无水四氢呋喃溶液(2.0mol/l,1.1l)中,温度控制在
‑
5~5℃。加完,继续保温
‑
5~5℃反应5h。然后,在
‑
5~5℃下减压除去溶剂和游离的二甲硫醚,直到体系无气泡为止。随后,补加入无水四氢呋喃(450ml)和(+)
‑
α
‑
蒎烯(70ml),继续在
‑
5~5℃反应3天,体系中产生大量白色固体。
[0090]
步骤二:制备硼酸化合物iii
‑1[0091]
将步骤一中的反应液降温至
‑
25~
‑
20℃后,于1h内滴加含有250.2克n
‑
boc
‑
吡咯啉的无水四氢呋喃(450ml)溶液,加完后保温
‑
25~
‑
20℃反应2天。缓慢滴加甲醇(500ml)淬灭反应,于2h内加完,保温
‑
10~0℃继续反应2h直到反应体系中固体完全消失,体系变澄清。
[0092]
后处理:减压浓缩得无色油状物,用甲醇(4l)和碳酸钾溶液(0.25g/ml,4l)溶解后,用石油醚(3l)萃取,分液,石油醚相再用甲醇(4l)和碳酸钾溶液(0.25g/ml,4l)洗涤,分液。水相合并后用石油醚(3l)萃取3次,分液。水相用盐酸酸化(ph1~2),加入氯化钠至饱和后,用乙酸乙酯(3l)萃取3次。合并的乙酸乙酯相用饱和食盐水洗涤,用无水硫酸钠干燥,过滤、浓缩后得化合物iii
‑
1的粗品527.8g,黄色油状物。
[0093]
步骤三:制备硼酸酯化合物iv
‑1[0094]
将步骤二中的黄色油状物溶于醋酸异丙酯(1l)中,加入230g频哪醇,室温反应2h。
反应液浓缩后通过快速硅胶柱层析纯化(淋洗剂为石油醚/乙酸乙酯,10/1),得450g化合物iv
‑
1,无色油状物。总收率为51%(按n
‑
boc
‑
吡咯啉计)。
[0095]1h nmr(400mhz,cdcl3)δ3.54
‑
3.49(m,2h),3.24
‑
3.18(m,2h),2.04
‑
1.95(m,1h),1.81
‑
1.70(m,1h),1.62
‑
1.50(m,1h),1.48(s,9h),1.22(s,12h).
[0096]
lcms:m/z=298.1;ee=98%。
[0097]
hplc:97.82%purity(220nm)。
[0098]
实施例2
[0099]
反应方程式如下:
[0100][0101]
制备方法同实施例1,将步骤三中的频哪醇替换为新戊二醇(192g)。得到366g化合物iv
‑
2,无色油状物。总收率为48%(按n
‑
boc
‑
吡咯啉计)。
[0102]1h nmr(400mhz,cdcl3)δ3.75(s,4h),3.55
‑
3.47(m,2h),3.24
‑
3.19(m,2h),2.05
‑
1.95(m,1h),1.80
‑
1.70(m,1h),1.60
‑
1.50(m,1h),1.48(s,9h),0.91(s,6h).
[0103]
lcms:m/z=284.1;ee=97%。
[0104]
hplc:98.50%purity(220nm)。
[0105]
实施例3
[0106]
反应方程式如下:
[0107][0108]
制备方法同实施例1,反应规模缩小5倍。步骤一中的(+)
‑
α
‑
蒎烯替换为(
‑
)
‑
α
‑
蒎烯;步骤三中的频哪醇替换为n
‑
甲基亚氨二乙酸(50g),醋酸异丙酯替换为甲苯,于110度反应24小时。化合物iv
‑
3的总收率为55%(按n
‑
boc
‑
吡咯啉计)。
[0109]1h nmr(400mhz,cdcl3)δ3.55
‑
3.47(m,2h),3.35(s,4h),3.24
‑
3.19(m,2h),2.32(s,3h),2.05
‑
1.95(m,1h),1.80
‑
1.70(m,1h),1.60
‑
1.50(m,1h),1.48(s,9h).
[0110]
lcms:m/z=327.1;ee=96%。
[0111]
hplc:99.10%purity(220nm)。
[0112]
实施例4
[0113]
反应方程式如下:
[0114][0115]
制备方法同实施例1,反应规模缩小5倍。步骤三中的频哪醇替换为l
‑
(+)
‑
酒石酸
二乙酯(55g)。化合物iv
‑
4的总收率为48%(按n
‑
boc
‑
吡咯啉计)。
[0116]1h nmr(400mhz,cdcl3)δ5.11(s,2h),4.25(q,j=8.0hz,4h),3.55
‑
3.47(m,2h),3.24
‑
3.19(m,2h),2.05
‑
1.95(m,1h),1.80
‑
1.70(m,1h),1.60
‑
1.50(m,1h),1.48(s,9h),1.22(t,j=8.0hz,6h).
[0117]
lcms:m/z=386.2;ee=97%。
[0118]
hplc:97.12%purity(220nm)。
[0119]
实施例5
[0120]
反应方程式如下:
[0121][0122]
制备方法同实施例1,反应规模缩小5倍。步骤三中的频哪醇替换为(1s,2s,3r,5s)
‑
(+)
‑
2,3
‑
蒎烷二醇(65g),化合物iv
‑
5的总收率为40%(按n
‑
boc
‑
吡咯啉计)。
[0123]1h nmr(400mhz,cdcl3)δ4.63(m,1h),3.84
‑
3.49(m,4h),3.24
‑
3.18(m,2h),2.04
‑
1.95(m,1h),1.81
‑
1.70(m,3h),1.65
‑
1.50(m,3h),1.48(s,9h),1.22(s,3h),0.88(s,6h).
[0124]
lcms:m/z=350.1;ee=96%。
[0125]
hplc:98.33%purity(220nm)。
[0126]
实施例6
[0127]
反应方程式如下:
[0128][0129]
制备方法同实施例1,反应规模缩小5倍。步骤三中的频哪醇替换为邻苯二酚(42g)。化合物iv
‑
6的总收率为45%(按n
‑
boc
‑
吡咯啉计)。
[0130]1h nmr(400mhz,cdcl3)δ6.77
‑
6.59(m,4h),3.54
‑
3.49(m,2h),3.24
‑
3.18(m,2h),2.04
‑
1.95(m,1h),1.81
‑
1.70(m,1h),1.62
‑
1.50(m,1h),1.48(s,9h).
[0131]
lcms:m/z=290.1;ee=97%。
[0132]
hplc:99.06%purity(220nm)。
[0133]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1