四氢吡咯并吡唑类衍生物、其制备方法及其在医药上的应用与流程
1.本公开属于医药领域,涉及一种通式(i)所示的四氢吡咯并吡唑类衍生物、其制备方法、含有该衍生物的药物组合物以及其作为治疗剂的用途,特别是作为akt1/2/3(akt pan)抑制剂的用途和在用于制备治疗和预防肿瘤的药物中的用途。
背景技术:
2.蛋白激酶b(pkb,又名akt)处于细胞中pi3k/akt/mtor信号传导的中心地位,它的功能对于细胞生长、存活、分化和代谢有着重要作用。pi3k信号通路参与及调控多个致癌基因和抗癌基因的表达,pi3k/akt信号通路的过度激活已经被证实与多种癌症的发生有关。
3.在细胞中,akt能够被一系列的信号激活,其中包括生长因子。当细胞膜上的受体酪氨酸激酶(receptor tyrosine kinase)被生长因子激活后,就会激活下游的pi3k,磷酸化
–
磷脂酰肌醇
‑
4,5
‑
二磷酸(phosphatidylinositol
‑
4,5
–
biphosphate,pip2)形成磷脂酰肌醇
‑
3,4,5
‑
三磷酸(phosphatidylinositol
‑
3,4,5
‑
triphosphate,pip3)。最后将磷脂酰肌醇依赖性激酶1(hosphatidylinositol
‑
dependent kinase 1,pdk1)和akt征召到细胞膜上,再由pdk1激活akt。pi3k的变异和pten的缺失、变异都会持续激活akt蛋白,令该通路被持续激活。akt在细胞内的作用主要是促进细胞增值,引起细胞从良性转化为恶性,推动细胞运动与侵袭,从而引起肿瘤细胞的转移与播散;而且高活性的磷酸化akt还会抑制细胞凋亡,并且参与化疗耐药的机制,影响临床治疗的效果。在临床统计中,具有高活性的akt的肿瘤在各个不同肿瘤中的占比均能达到40%或以上。
4.akt酶有3个亚型(akt1,akt2和akt3),在各项研究中显示,它们各自在体内有着不同的功能。akt1激活的信号通路主要调控细胞的增殖和存活,akt2则参与细胞侵袭和迁移,以及胰岛素调控的血糖代谢通路等功能。akt3的基因敲除老鼠虽然只显示与胚胎大脑发育相关的功能,但是在临床研究中发现akt3的表达量在乳腺癌等多种肿瘤中有明显上升。此外在临床前的体外研究中显示,乳腺癌细胞在长期akt1/2选择性抑制剂mk2206的处理中,会产生耐药性,而akt3的表达量在该耐药细胞中明显上升。
5.针对akt靶点的抑制剂,在临床上已经研究多年。akt1/2的选择性抑制剂mk2206(merck)和bay1125976(bayer)在临床上因疗效和毒性等原因并没有取得成功。然而近年,akt1/2/3(akt pan)抑制剂azd5363(az)和gdc0068(roche)在临床2期取得突破性结果,它们和其他抗癌药物的联用对三阴性乳腺癌,er+乳腺癌和前列腺癌的治疗产生明显的疗效。目前这两款akt1/2/3(akt pan)抑制剂azd5363和gdc0068已经成功的推进3期临床阶段。
6.2018年的全球癌症统计数字显示,全球有1800万新增癌症病例和960万人的癌症死亡案例,每年癌症发病率均呈上升趋势。其中排名前三的癌症分别是肺癌(11.6%),女性乳腺癌(11.6%),前列腺癌(7.1%)。在中国,由于我国人口基数庞大,女性乳腺癌发病例数和死亡例数分别占全球发病和死亡的11.2%和9.2%,在世界范围内位居前列;前列腺癌在美国则属于的高发癌症,预计2022年全球前列腺癌患者会达到1100万,其中美国约300万(28%)。
7.已经公开的akt抑制剂的专利申请包括wo2006/071819、us8377937、wo2008/075109、us2010120801和wo2009006040。
技术实现要素:
8.本公开的目的在于提供一种通式(i)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐:
[0009][0010]
其中:
[0011]
环中的表示单键或双键;
[0012]
y和z选自氮原子或碳原子;
[0013]
r0选自
‑
c(o)chr5r6或
‑
c(o)nhchr5r6;
[0014]
r1相同或不同,各自独立地选自氢原子、卤素、烷基、羟烷基、烷氧基、卤代烷氧基和卤代烷基;
[0015]
r2相同或不同,各自独立地选自氢原子、氧代、卤素、烷基、烯基、炔基、烷氧基、卤代烷基、卤代烷氧基、氰基、
‑
nr7r8、硝基、羟基、羟烷基、环烷基、杂环基、芳基和杂芳基,其中所述的烷基任选被选自
‑
nr7r8、卤素、烷氧基、卤代烷基、卤代烷氧基、氰基、硝基、羟基、羟烷基、环烷基、杂环基、芳基和杂芳基中的一个或多个取代基所取代;或者两个r2与所连接的同一碳原子可形成环烷基;
[0016]
r3和r4相同或不同,各自独立地选自氢原子、卤素、烷基、烯基、炔基、烷氧基、卤代烷基、卤代烷氧基、氰基、氨基、硝基、羟基、羟烷基、环烷基、杂环基、芳基和杂芳基;
[0017]
r5选自卤素、烷基、烯基、炔基、烷氧基、卤代烷基、卤代烷氧基、氰基、氨基、硝基、羟基、羟烷基、环烷基、杂环基、芳基和杂芳基,其中所述的烷基、烯基、炔基、烷氧基、羟烷基、环烷基、杂环基、芳基和杂芳基各自独立地任选被选自
‑
nr9r
10
、氧代、卤素、烷基、烯基、炔基、烷氧基、卤代烷基、卤代烷氧基、氰基、硝基、羟基、羟烷基、环烷基、杂环基、芳基和杂芳基中的一个或多个取代基所取代;
[0018]
r6选自卤素、烷基、烯基、炔基、烷氧基、卤代烷基、卤代烷氧基、氰基、氨基、硝基、羟基、羟烷基、环烷基、杂环基、芳基和杂芳基,其中所述的烷基、烯基、炔基、烷氧基、羟烷基、环烷基、杂环基、芳基和杂芳基各自独立地任选被选自氧代、卤素、烷基、烯基、炔基、烷氧基、卤代烷基、卤代烷氧基、氰基、氨基、硝基、羟基、羟烷基、环烷基、杂环基、芳基和杂芳基中的一个或多个取代基所取代;
[0019]
r7和r8相同或不同,各自独立地选自氢原子、卤素、烷基、烯基、炔基、烷氧基、卤代烷基、卤代烷氧基、氰基、氨基、硝基、羟基、羟烷基、环烷基、杂环基、芳基和杂芳基;
[0020]
r9和r
10
相同或不同,各自独立地选自氢原子、卤素、烷基、烯基、炔基、烷氧基、卤代烷基、卤代烷氧基、氰基、氨基、硝基、羟基、羟烷基、环烷基、环烷基烷基、杂环基、芳基和杂芳基;
[0021]
n为0、1、2、3或4;
[0022]
q为0、1、2、3、4或5。
[0023]
在本公开一些优选的实施方案中,所述的通式(i)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中y为碳原子。
[0024]
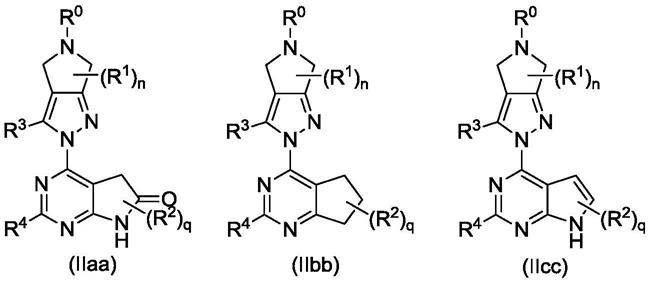
在本公开一些优选的实施方案中,所述的通式(i)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其为通式(iiaa)、(iibb)或(iicc)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐:
[0025][0026]
其中:
[0027]
r0‑
r4、n和q如通式(i)中所定义。
[0028]
在本公开一些优选的实施方案中,所述的通式(i)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其为通式(iiiaa)或通式(iiibb)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐:
[0029][0030]
其中:
[0031]
环中的表示单键或双键;
[0032]
r0‑
r4、r6、r9、y、q和n如通式(i)中所定义。
[0033]
在本公开一些优选的实施方案中,所述的通式(i)、(iiaa)、(iibb)、(iicc)或(iiibb)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中r0为
‑
c(o)chr5r6,r5、r6如通式(i)中所定义;
[0034]
优选地,r0为r6、r9和r
10
如通式(i)、(iiaa)、(iibb)、(iicc)或(iiibb)中所定义,其中为
[0035]
在本公开的一些实施方案中,所述的通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)或(iiibb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中r5为烷基,其中所述的烷基任选被选自羟基和
‑
nr9r
10
中的一个或多个取代基所取代;r9和r
10
相同或不同,各自独立地选自氢原子或烷基;r6选自芳基或杂芳基,其中所述的芳基或杂芳基任选被选自卤素、烷基、烷氧基、卤代烷基和卤代烷氧基中的一个或多个取代基所取代;
[0036]
优选地,r5为c1‑6烷基,其中所述的c1‑6烷基任选被选自羟基和
‑
nr9r
10
中的一个或多个取代基所取代;r9和r
10
相同或不同,各自独立地选自氢原子或c1‑6烷基;r6选自c6‑
10
芳基或5
‑
10元杂芳基,其中所述的c6‑
10
芳基或5
‑
10元杂芳基任选被选自卤素、c1‑6烷基、c1‑6烷氧基、c1‑6卤代烷基和c1‑6卤代烷氧基中的一个或多个取代基所取代。
[0037]
在本公开的一些实施方案中,所述的通式(i)、(iiaa)、(iibb)、(iicc)或(iiibb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中r5为被一个或多个,优选一个
‑
nr9r
10
取代的c1‑6烷基,r9和r
10
相同或不同,各自独立地选自氢原子或c1‑6烷基;优选地,r5为被
‑
nr9r
10
取代的甲基,r9和r
10
相同或不同,各自独立地选自氢原子或异丙基。
[0038]
在本公开的一些实施方案中,所述的通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)或(iiibb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中r6为c6‑
10
芳基,优选苯基,其任选被一个或多个卤素取代。
[0039]
在本公开的一些实施方案中,所述的通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)或(iiibb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中r1选自氢原子、c1‑6烷基和c1‑6羟烷基;优选地,r1选自氢原子、甲基和羟甲基;更优选地,r1为氢原子。
[0040]
在本公开的一些实施方案中,所述的通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)或(iiibb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中r2相同或不同,各自独立地选自氢原子、氧代、卤素、烷基、卤代烷基、羟烷基、羟基、烷氧基和卤代烷氧基;或者两个r2与所连接的同一碳原子可
为r6选自c6‑
10
芳基或5
‑
10元杂芳基,其中所述的c6‑
10
芳基或5
‑
10元杂芳基任选被选自卤素、c1‑6烷基、c1‑6烷氧基、c1‑6卤代烷基和c1‑6卤代烷氧基中的一个或多个取代基所取代;r9和r
10
相同或不同,各自独立地选自氢原子或c1‑6烷基。
[0050]
在本公开的一些实施方案中,所述的通式(iicc)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中r2相同或不同,各自独立地选自氢原子和c1‑6烷基;q为0、1或2;r3选自氢原子、卤素、c1‑6烷基和c1‑6卤代烷基;r4选自氢原子、卤素、c1‑6烷基和c1‑6卤代烷基;r1选自氢原子、c1‑6烷基和c1‑6羟烷基;n为0或1;r0为r6选自c6‑
10
芳基或5
‑
10元杂芳基,其中所述的c6‑
10
芳基或5
‑
10元杂芳基任选被选自卤素、c1‑6烷基、c1‑6烷氧基、c1‑6卤代烷基和c1‑6卤代烷氧基中的一个或多个取代基所取代;r9和r
10
相同或不同,各自独立地选自氢原子或c1‑6烷基。
[0051]
在本公开的一些实施方案中,所述的通式(iiaa)、(iicc)或(iiiaa)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中q为0、1、2或3;优选为0、1或2。
[0052]
在本公开的一些实施方案中,所述的通式(iibb)或(iiibb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中q为0、1、2、3、4或5。
[0053]
表a本公开的典型化合物包括但不限于:
[0054]
[0055]
[0056]
[0057]
[0058]
[0059][0060]
本公开的另一方面涉及通式(iiiaaa)或(iiiabb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其盐,
[0061][0062]
其中:
[0063]
环中的表示单键或双键;
[0064]
r
w
、r
m
为氨基保护基;
[0065]
r
n
为羟基保护基;
[0066]
r0‑
r4、r6、r9、y、n和q如通式(iiiaa)或(iiibb)中所定义。
[0067]
本公开的另一方面涉及通式(iiiaaa)或(iiiabb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其盐,其中r
m
为叔丁氧羰基,r
w
为(三甲基硅)乙氧基甲基,r
n
为对硝基苯甲酰基。
[0068]
在本公开的一些实施方案中,所述的通式(iiiaaa)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中q为0、1或2。
[0069]
在本公开的一些实施方案中,所述的通式(iiiabb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中q为0、1、2、3、4或5。
[0070]
本公开的典型中间体化合物包括但不限于:
[0071]
[0072]
[0073]
[0074][0075]
其中boc为叔丁氧羰基;sem为(三甲基硅)乙氧基甲基。
[0076]
本公开的另一方面涉及一种制备通式(iiiaa)或通式(iiibb)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐的方法,该方法包括:
[0077][0078]
通式(iiiaaa)的化合物或其盐脱去氨基保护基,得到通式(iiiaa)的化合物,或
[0079]
通式(iiiabb)的化合物或其盐脱去羟基保护基,得到通式(iiibb)的化合物,
[0080]
其中:
[0081]
环中的表示单键或双键;
[0082]
r
w
、r
m
为氨基保护基;
[0083]
r
n
为羟基保护基;
[0084]
r0‑
r4、r6、r9、y、n和q如通式(iiiaa)或(iiibb)中所定义。
[0085]
本公开的另一方面涉及一种药物组合物,所述药物组合物含有本公开通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,以及一种或多种药学上可接受的载体、稀释剂或赋形剂。
[0086]
本公开进一步涉及通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,或包含其的药物组合物在制备用于抑制akt1/2/3(akt pan)的药物中的用途。
[0087]
本公开进一步涉及通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,或包含其的药物组合物在制备用于治疗和/或预防肿瘤的药物中的用途;优选地,在制备用于治疗和/或预防癌症的药物中的用途。
[0088]
本公开进一步涉及通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示
的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,或包含其的药物组合物在制备用于治疗和/或预防疾病或病症的药物中的用途;其中所述的疾病或病症优选选自卵巢癌、乳腺癌、前列腺癌、神经胶质瘤、胶质细胞瘤、胃癌、输卵管癌、肺癌、腹膜肿瘤、黑色素瘤、脑癌、食管癌、肝癌、胰腺癌、结肠直肠癌、肺癌、肾癌、宫颈癌、皮肤癌、神经母细胞瘤、肉瘤、骨癌、子宫癌、子宫内膜癌、头颈肿瘤、多发性骨髓瘤、淋巴瘤、非霍奇金淋巴瘤、非小细胞肺癌、真性红细胞增多症、白血病、甲状腺肿瘤、膀胱癌和胆囊癌。
[0089]
本公开还涉及一种抑制akt1/2/3(akt pan)的方法,其包括给予所需患者治疗有效量的通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,或包含其的药物组合物。
[0090]
本公开还涉及一种治疗和/或预防肿瘤的方法,优选地,治疗和/或预防癌症的方法,其包括给予所需患者治疗有效量的通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,或包含其的药物组合物。
[0091]
本公开还涉及一种治疗和/或预防疾病或病症的方法,其包括给予所需患者治疗有效量的通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,或包含其的药物组合物,其中所述的疾病或病症优选选自卵巢癌、乳腺癌、前列腺癌、神经胶质瘤、胶质细胞瘤、胃癌、输卵管癌、肺癌、腹膜肿瘤、黑色素瘤、脑癌、食管癌、肝癌、胰腺癌、结肠直肠癌、肺癌、肾癌、宫颈癌、皮肤癌、神经母细胞瘤、肉瘤、骨癌、子宫癌、子宫内膜癌、头颈肿瘤、多发性骨髓瘤、淋巴瘤、非霍奇金淋巴瘤、非小细胞肺癌、真性红细胞增多症、白血病、甲状腺肿瘤、膀胱癌和胆囊癌。
[0092]
本公开进一步涉及一种通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式、或其可药用盐或包含其的药物组合物,其用作药物。
[0093]
本公开还涉及通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,或包含其的药物组合物,其用作akt1/2/3(akt pan)抑制剂。
[0094]
本公开进一步涉及通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,或包含其的药物组合物,其用于治疗肿瘤;优选地,用于治疗癌症。
[0095]
本公开中所述的肿瘤选自黑色素瘤、脑瘤、食管癌、胃癌、肝癌、胰腺癌、结肠直肠癌、肺癌、肾癌、乳腺癌、宫颈癌、卵巢癌、前列腺癌、皮肤癌、神经母细胞瘤、神经胶质瘤、胶质细胞瘤、肉瘤、骨癌、子宫癌、子宫内膜癌、头颈肿瘤、多发性骨髓瘤、b
‑
细胞淋巴瘤、真性红细胞增多症、白血病、甲状腺肿瘤、膀胱癌和胆囊癌。
[0096]
本公开进一步涉及通式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其可药用盐,或包含其的药物组合物,其用于治疗疾病或病症;其中所述的疾病或病
症优选选自卵巢癌、乳腺癌、前列腺癌、神经胶质瘤、胶质细胞瘤、胃癌、输卵管癌、肺癌、腹膜肿瘤、黑色素瘤、脑癌、食管癌、肝癌、胰腺癌、结肠直肠癌、肺癌、肾癌、宫颈癌、皮肤癌、神经母细胞瘤、肉瘤、骨癌、子宫癌、子宫内膜癌、头颈肿瘤、多发性骨髓瘤、淋巴瘤、非霍奇金淋巴瘤、非小细胞肺癌、真性红细胞增多症、白血病、甲状腺肿瘤、膀胱癌和胆囊癌。
[0097]
以上所述的肿瘤、癌症、疾病或病症优选为由akt1/2/3介导的肿瘤、癌症、疾病或病症。
[0098]
可将活性化合物制成适合于通过任何适当途径给药的形式,通过常规方法使用一种或多种药学上可接受的载体来配制本公开的组合物。因此,本公开的活性化合物可以配制成用于口服给药、注射(例如静脉内、肌肉内或皮下)给药,吸入或吹入给药的各种剂型。本公开的化合物也可以配制成持续释放剂型,例如片剂、硬或软胶囊、水性或油性混悬液、乳剂、注射液、可分散性粉末或颗粒、栓剂、锭剂或糖浆。
[0099]
本公开治疗方法中所用化合物或组合物的剂量通常将随疾病的严重性、患者的体重和化合物的相对功效而改变。不过,作为一般性指导,活性化合物优选是以单位剂量的方式,或者是以患者可以以单剂自我给药的方式。本公开化合物或组合物的单位剂量的表达方式可以是片剂、胶囊、扁囊剂、瓶装药水、药粉、颗粒剂、锭剂、栓剂、再生药粉或液体制剂。合适的单位剂量可以是0.1~1000mg。
[0100]
本公开的药物组合物除活性化合物外,可含有一种或多种辅料,所述辅料选自以下成分:填充剂(稀释剂)、粘合剂、润湿剂、崩解剂或赋形剂等。根据给药方法的不同,组合物可含有0.1至99重量%的活性化合物。
[0101]
片剂含有活性成分和用于混合的适宜制备片剂的无毒的可药用的赋形剂。这些赋形剂可以是惰性赋形剂、造粒剂、崩解剂、粘合剂和润滑剂。这些片剂可以不包衣或可通过掩盖药物的味道或在胃肠道中延迟崩解和吸收,因而在较长时间内提供缓释作用的已知技术将其包衣。
[0102]
也可用其中活性成分与惰性固体稀释剂或其中活性成分与水溶性载体或油溶媒混合的软明胶胶囊提供口服制剂。
[0103]
水混悬液含有活性物质和用于混合的适宜制备水悬浮液的赋形剂。此类赋形剂是悬浮剂、分散剂或湿润剂。水混悬液也可以含有一种或多种防腐剂、一种或多种着色剂、一种或多种矫味剂和一种或多种甜味剂。
[0104]
油混悬液可通过使活性成分悬浮于植物油,或矿物油配制而成。油悬浮液可含有增稠剂。可加入上述的甜味剂和矫味剂,以提供可口的制剂。可通过加入抗氧化剂保存这些组合物。
[0105]
本公开的药物组合物也可以是水包油乳剂的形式。油相可以是植物油,或矿物油或其混合物。适宜的乳化剂可以是天然产生的磷脂,乳剂也可以含有甜味剂、矫味剂、防腐剂和抗氧剂。此类制剂也可含有缓和剂、防腐剂、着色剂和抗氧剂。
[0106]
本公开的药物组合物可以是无菌注射水溶液形式。可以使用的可接受的溶媒或溶剂有水、林格氏液和等渗氯化钠溶液。无菌注射制剂可以是其中活性成分溶于油相的无菌注射水包油微乳可通过局部大量注射,将注射液或微乳注入患者的血流中。或者,最好按可保持本公开化合物恒定循环浓度的方式给予溶液和微乳。为保持这种恒定浓度,可使用连续静脉内递药装置。这种装置的实例是deltec cadd
‑
plus.tm.5400型静脉注射泵。
[0107]
本公开的药物组合物可以是用于肌内和皮下给药的无菌注射水或油混悬液的形式。可按已知技术,用上述那些适宜的分散剂或湿润剂和悬浮剂配制该混悬液。无菌注射制剂也可以是在肠胃外可接受的无毒稀释剂或溶剂中制备的无菌注射溶液或混悬液。此外,可方便地用无菌固定油作为溶剂或悬浮介质。为此目的,可使用任何调和固定油。此外,脂肪酸也可以制备注射剂。
[0108]
可按用于直肠给药的栓剂形式给予本公开化合物。可通过将药物与在普通温度下为固体但在直肠中为液体,因而在直肠中会溶化而释放药物的适宜的无刺激性赋形剂混合来制备这些药物组合物。
[0109]
可通过加入水来制备水混悬的可分散粉末和颗粒给予本公开化合物。可通过将活性成分与分散剂或湿润剂、悬浮剂或一种或多种防腐剂混合来制备这些药物组合物。
[0110]
如本领域技术人员所熟知的,药物的给药剂量依赖于多种因素,包括但并非限定于以下因素:所用具体化合物的活性、患者的年龄、患者的体重、患者的健康状况、患者的行为、患者的饮食、给药时间、给药方式、排泄的速率、药物的组合等;另外,最佳的治疗方式如治疗的模式、化合物的日用量或可药用的盐的种类可以根据传统的治疗方案来验证。
[0111]
发明的详细说明
[0112]
除非有相反陈述,在说明书和权利要求书中使用的术语具有下述含义。
[0113]
术语“烷基”指饱和脂肪族烃基团,其为包含1至20个碳原子的直链或支链基团,优选含有1至12个(例如1、2、3、4、5、6、7、8、9、10、11和12个)碳原子的烷基,更优选为含有1至6个碳原子的烷基。非限制性实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1
‑
二甲基丙基、1,2
‑
二甲基丙基、2,2
‑
二甲基丙基、1
‑
乙基丙基、2
‑
甲基丁基、3
‑
甲基丁基、正己基、1
‑
乙基
‑2‑
甲基丙基、1,1,2
‑
三甲基丙基、1,1
‑
二甲基丁基、1,2
‑
二甲基丁基、2,2
‑
二甲基丁基、1,3
‑
二甲基丁基、2
‑
乙基丁基、2
‑
甲基戊基、3
‑
甲基戊基、4
‑
甲基戊基、2,3
‑
二甲基丁基、正庚基、2
‑
甲基己基、3
‑
甲基己基、4
‑
甲基己基、5
‑
甲基己基、2,3
‑
二甲基戊基、2,4
‑
二甲基戊基、2,2
‑
二甲基戊基、3,3
‑
二甲基戊基、2
‑
乙基戊基、3
‑
乙基戊基、正辛基、2,3
‑
二甲基己基、2,4
‑
二甲基己基、2,5
‑
二甲基己基、2,2
‑
二甲基己基、3,3
‑
二甲基己基、4,4
‑
二甲基己基、2
‑
乙基己基、3
‑
乙基己基、4
‑
乙基己基、2
‑
甲基
‑2‑
乙基戊基、2
‑
甲基
‑3‑
乙基戊基、正壬基、2
‑
甲基
‑2‑
乙基己基、2
‑
甲基
‑3‑
乙基己基、2,2
‑
二乙基戊基、正癸基、3,3
‑
二乙基己基、2,2
‑
二乙基己基,及其各种支链异构体等。更优选的是含有1至6个碳原子的低级烷基,非限制性实施例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1
‑
二甲基丙基、1,2
‑
二甲基丙基、2,2
‑
二甲基丙基、1
‑
乙基丙基、2
‑
甲基丁基、3
‑
甲基丁基、正己基、1
‑
乙基
‑2‑
甲基丙基、1,1,2
‑
三甲基丙基、1,1
‑
二甲基丁基、1,2
‑
二甲基丁基、2,2
‑
二甲基丁基、1,3
‑
二甲基丁基、2
‑
乙基丁基、2
‑
甲基戊基、3
‑
甲基戊基、4
‑
甲基戊基、2,3
‑
二甲基丁基等。烷基可以是取代的或非取代的,当被取代时,取代基可以在任何可使用的连接点上被取代,所述取代基优选独立地任选选自d原子、卤素、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基、杂芳基中的一个或多个取代基。
[0114]
术语“亚烷基”指饱和的直链或支链脂肪族烃基,其为从母体烷的相同碳原子或两个不同的碳原子上除去两个氢原子所衍生的残基,其为包含1至20个碳原子的直链或支链基团,优选含有1至12个(例如1、2、3、4、5、6、7、8、9、10、11和12个)碳原子,更优选含有1至6
个碳原子的亚烷基。亚烷基的非限制性实例包括但不限于亚甲基(
‑
ch2‑
)、1,1
‑
亚乙基(
‑
ch(ch3)
‑
)、1,2
‑
亚乙基(
‑
ch2ch2)
‑
、1,1
‑
亚丙基(
‑
ch(ch2ch3)
‑
)、1,2
‑
亚丙基(
‑
ch2ch(ch3)
‑
)、1,3
‑
亚丙基(
‑
ch2ch2ch2‑
)、1,4
‑
亚丁基(
‑
ch2ch2ch2ch2‑
)等。亚烷基可以是取代的或非取代的,当被取代时,取代基可以在任何可使用的连接点上被取代,所述取代基优选独立地任选选自烯基、炔基、烷氧基、卤代烷氧基、环烷基氧基、杂环基氧基、烷硫基、烷基氨基、卤素、巯基、羟基、硝基、氰基、环烷基、杂环基、芳基、杂芳基、环烷氧基、杂环烷氧基、环烷硫基、杂环烷硫基和氧代基中的一个或多个取代基。
[0115]
术语“烯基”指分子中含有碳碳双键的烷基化合物,其中烷基的定义如上所述。烯基可以是取代的或非取代的,当被取代时,取代基优选为一个或多个以下基团,其独立地选自烷氧基、卤素、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基和杂芳基。
[0116]
术语“炔基”指分子中含有碳碳三键的烷基化合物,其中烷基的定义如上所述。炔基可以是取代的或非取代的,当被取代时,取代基优选为一个或多个以下基团,其独立地选自烷氧基、卤素、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基和杂芳基。
[0117]
术语“环烷基”指饱和或部分不饱和单环或多环环状烃取代基,环烷基环包含3至20个碳原子,优选包含3至12个碳原子,优选包含3至8个(例如3、4、5、6、7和8个)碳原子,更优选包含3至6个碳原子。单环环烷基的非限制性实例包括环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环己二烯基、环庚基、环庚三烯基、环辛基等;多环环烷基包括螺环、稠环和桥环的环烷基。
[0118]
术语“螺环烷基”指5至20元,单环之间共用一个碳原子(称螺原子)的多环基团,其可以含有一个或多个双键。优选为6至14元,更优选为7至10元(例如7、8、9或10元)。根据环与环之间共用螺原子的数目将螺环烷基分为单螺环烷基、双螺环烷基或多螺环烷基,优选为单螺环烷基和双螺环烷基。更优选为3元/5元、3元/6元、4元/4元、4元/5元、4元/6元、5元/5元或5元/6元单螺环烷基。螺环烷基的非限制性实例包括:
[0119][0120]
术语“稠环烷基”指5至20元,系统中的每个环与体系中的其他环共享毗邻的一对碳原子的全碳多环基团,其中一个或多个环可以含有一个或多个双键。优选为6至14元,更优选为7至10元(例如7、8、9或10元)。根据组成环的数目可以分为双环、三环、四环或多环稠环烷基,优选为双环或三环,更优选为3元/4元、3元/5元、3元/6元、4元/4元、4元/5元、4元/6元、5元/4元、5元/5元、5元/6元、6元/3元、6元/4元、6元/5元和6元/6元的双环烷基。稠环烷基的非限制性实例包括:
[0121]
[0122]
术语“桥环烷基”指5至20元,任意两个环共用两个不直接连接的碳原子的全碳多环基团,其可以含有一个或多个双键。优选为6至14元,更优选为7至10元(例如7、8、9或10元)。根据组成环的数目可以分为双环、三环、四环或多环桥环烷基,优选为双环、三环或四环,更优选为双环或三环。桥环烷基的非限制性实例包括:
[0123][0124]
所述环烷基环包括如上所述的环烷基(包括单环、螺环、稠环和桥环)稠合于芳基、杂芳基或杂环烷基环上,其中与母体结构连接在一起的环为环烷基,非限制性实例包括茚满基、四氢萘基、苯并环庚烷基等;优选茚满基、四氢萘基。
[0125]
环烷基可以是取代的或非取代的,当被取代时,取代基可以在任何可使用的连接点上被取代,所述取代基优选独立地任选选自卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基、杂芳基中的一个或多个取代基。
[0126]
术语“烷氧基”指
‑
o
‑
(烷基),其中烷基的定义如上所述。烷氧基的非限制性实例包括:甲氧基、乙氧基、丙氧基和丁氧基。烷氧基可以是任选取代的或非取代的,当被取代时,取代基优选为一个或多个以下基团,其独立地选自d原子、卤素、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基和杂芳基。
[0127]
术语“杂环基”指饱和或部分不饱和单环或多环环状烃取代基,其包含3至20个环原子,其中一个或多个环原子为选自氮、氧、硫、s(o)或s(o)2的杂原子,但不包括
‑
o
‑
o
‑
、
‑
o
‑
s
‑
或
‑
s
‑
s
‑
的环部分,其余环原子为碳。优选包含3至12个(例如3、4、5、6、7、8、9、10、11和12个)环原子,其中1~4个(例如1、2、3和4个)是杂原子;更优选包含3至8个环原子(例如3、4、5、6、7和8个),其中1
‑
3个(例如1、2和3个)是杂原子;更优选包含3至6个环原子,其中1
‑
3个是杂原子;最优选包含5或6个环原子,其中1
‑
3个是杂原子。单环杂环基的非限制性实例包括吡咯烷基、四氢吡喃基、1,2.3.6
‑
四氢吡啶基、哌啶基、哌嗪基、吗啉基、硫代吗啉基、高哌嗪基等。多环杂环基包括螺环、稠环和桥环的杂环基。
[0128]
术语“螺杂环基”指5至20元,单环之间共用一个原子(称螺原子)的多环杂环基团,其中一个或多个环原子为选自氮、氧、硫、s(o)或s(o)2的杂原子,其余环原子为碳。其可以含有一个或多个双键。优选为6至14元,更优选为7至10元(例如7、8、9或10元)。根据环与环之间共用螺原子的数目将螺杂环基分为单螺杂环基、双螺杂环基或多螺杂环基,优选为单螺杂环基和双螺杂环基。更优选为3元/5元、3元/6元、4元/4元、4元/5元、4元/6元、5元/5元或5元/6元单螺杂环基。螺杂环基的非限制性实例包括:
[0129][0130]
术语“稠杂环基”指5至20元,系统中的每个环与体系中的其他环共享毗邻的一对原子的多环杂环基团,一个或多个环可以含有一个或多个双键,其中一个或多个环原子为选自氮、氧、硫、s(o)或s(o)2的杂原子,其余环原子为碳。优选为6至14元,更优选为7至10元(例如7、8、9或10元)。根据组成环的数目可以分为双环、三环、四环或多环稠杂环基,优选为双环或三环,更优选为3元/4元、3元/5元、3元/6元、4元/4元、4元/5元、4元/6元、5元/4元、5元/5元、5元/6元、6元/3元、6元/4元、6元/5元和6元/6元双环稠杂环基。稠杂环基的非限制性实例包括:
[0131][0132]
术语“桥杂环基”指5至14元,任意两个环共用两个不直接连接的原子的多环杂环基团,其可以含有一个或多个双键,其中一个或多个环原子为选自氮、氧、硫、s(o)或s(o)2的杂原子,其余环原子为碳。优选为6至14元,更优选为7至10元(例如7、8、9或10元)。根据组成环的数目可以分为双环、三环、四环或多环桥杂环基,优选为双环、三环或四环,更优选为双环或三环。桥杂环基的非限制性实例包括:
[0133][0134]
所述杂环基环包括如上所述的杂环基(包括单环、螺杂环、稠杂环和桥杂环)稠合于芳基、杂芳基或环烷基环上,其中与母体结构连接在一起的环为杂环基,其非限制性实例包括:
[0135]
等。
[0136]
杂环基可以是取代的或非取代的,当被取代时,取代基可以在任何可使用的连接点上被取代,所述取代基优选独立地任选选自卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、
环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基、杂芳基中的一个或多个取代基。
[0137]
术语“芳基”指具有共轭的π电子体系的6至14元全碳单环或稠合多环(稠合多环是共享毗邻碳原子对的环)基团,优选为6至10元,例如苯基和萘基。所述芳基环包括如上所述的芳基环稠合于杂芳基、杂环基或环烷基环上,其中与母体结构连接在一起的环为芳基环,其非限制性实例包括:
[0138][0139]
芳基可以是取代的或非取代的,当被取代时,取代基可以在任何可使用的连接点上被取代,所述取代基优选独立地任选选自卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基、杂芳基中的一个或多个取代基。
[0140]
术语“杂芳基”指包含1至4个(例如1、2、3和4个)杂原子、5至14个环原子的杂芳族体系,其中杂原子选自氧、硫和氮。杂芳基优选为5至10元(例如5、6、7、8、9或10元),更优选为5元或6元,例如呋喃基、噻吩基、吡啶基、吡咯基、n
‑
烷基吡咯基、嘧啶基、吡嗪基、哒嗪基、咪唑基、吡唑基、三唑基、四唑基等。所述杂芳基环包括如上述的杂芳基稠合于芳基、杂环基或环烷基环上,其中与母体结构连接在一起的环为杂芳基环,其非限制性实例包括:
[0141][0142]
杂芳基可以是取代的或非取代的,当被取代时,取代基可以在任何可使用的连接点上被取代,所述取代基优选独立地任选选自卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基、杂芳基中的一个或多个取代基。
[0143]
上述环烷基、杂环基、芳基和杂芳基包括从母体环原子上除去一个氢原子所衍生的残基,或从母体的相同或两个不同的环原子上除去两个氢原子所衍生的残基,即“二价环烷基”、“二价杂环基”、“亚芳基”、“亚杂芳基”。
[0144]
术语“氨基保护基”是为了使分子其它部位进行反应时氨基保持不变,用易于脱去的基团对氨基进行保护。非限制性实施例包含(三甲基硅)乙氧基甲基、四氢吡喃基、叔丁氧羰基、乙酰基、苄基、烯丙基和对甲氧苄基等。这些基团可任选地被选自卤素、烷氧基或硝基中的1
‑
3个取代基所取代。所述氨基保护基优选为(三甲基硅)乙氧基甲基和叔丁氧羰基。
[0145]
术语“羟基保护基”是本领域已知的适当的用于羟基保护的基团,参见文献(“protective groups in organic synthesis”,5
th ed.t.w.greene&p.g.m.wuts)中的羟基保护基团。作为示例,优选地,所述的羟基保护基可以是(c1‑
10
烷基或芳基)3硅烷基,例如:三乙基硅基,三异丙基硅基,叔丁基二甲基硅基,叔丁基二苯基硅基等;可以是c1‑
10
烷基或取代烷基,优选烷氧基或芳基取代的烷基,更优选c1‑6烷氧基取代的c1‑6烷基或苯基取代的c1‑6烷基,最优选c1‑4烷氧基取代的c1‑4烷基,例如:甲基,叔丁基,烯丙基,苄基,甲氧基甲基(mom),乙氧基乙基,2
‑
四氢吡喃基(thp)等;可以是(c1‑
10
烷基或芳香基)酰基,例如:甲酰基,乙酰基,苯甲酰基、对硝基苯甲酰基等;可以是(c1‑6烷基或c6‑
10
芳基)磺酰基;也可以是(c1‑6烷氧基或c6‑
10
芳基氧基)羰基。所述羟基保护基优选为对硝基苯甲酰基。
[0146]
术语“环烷基氧基”指环烷基
‑
o
‑
,其中环烷基如上所定义。
[0147]
术语“杂环基氧基”指杂环基
‑
o
‑
,其中杂环基如上所定义。
[0148]
术语“烷硫基”指烷基
‑
s
‑
,其中烷基如上所定义。
[0149]
术语“卤代烷基”指烷基被一个或多个卤素取代,其中烷基如上所定义。
[0150]
术语“卤代烷氧基”指烷氧基被一个或多个卤素取代,其中烷氧基如上所定义。
[0151]
术语“氘代烷基”指烷基被一个或多个氘原子取代,其中烷基如上所定义。
[0152]
术语“羟烷基”指被一个或多个羟基取代的烷基,其中烷基如上所定义。
[0153]
术语“卤素”指氟、氯、溴或碘。
[0154]
术语“羟基”指
‑
oh。
[0155]
术语“巯基”指
‑
sh。
[0156]
术语“氨基”指
‑
nh2。
[0157]
术语“氰基”指
‑
cn。
[0158]
术语“硝基”指
‑
no2。
[0159]
术语“氧代”指“=o”。
[0160]
术语“羰基”指c=o。
[0161]
术语“羧基”指
‑
c(o)oh。
[0162]
术语“羧酸酯基”指
‑
c(o)o(烷基)、
‑
c(o)o(环烷基)、(烷基)c(o)o
‑
或(环烷基)c(o)o
‑
,其中烷基和环烷基如上所定义。
[0163]
boc为叔丁氧羰基;sem为(三甲基硅基)乙氧基甲基。
[0164]
本公开还包括各种氘化形式的式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a化合物。与碳原子连接的各个可用的氢原子可独立地被氘原子替换。本领域技术人员能够参考相关文献合成氘化形式的式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a化合物。在制备氘代形式的式(i)、(iiaa)、(iibb)、(iicc)、(iiiaa)、(iiibb)或表a化合物时可使用市售的氘代起始物质,或它们可使用常规技术采用氘代试剂合成,氘代试剂包括但不限于氘代硼烷、三氘代硼烷四氢呋喃溶液、氘代氢化锂铝、氘代碘乙烷和氘代碘甲烷等。氘代物通常可以保留与未氘代的化合物相当的活性,并且当氘代在某些特定位点时可以取得更好的代谢稳定性,从而获得某些治疗优势。
[0165]“任选”或“任选地”意味着随后所描述的事件或环境可以但不必发生,该说明包括该事件或环境发生或不发生地场合。例如,“任选被烷基取代的杂环基团”意味着烷基可以但不必须存在,该说明包括杂环基团被烷基取代的情形和杂环基团不被烷基取代的情形。
[0166]“取代的”指基团中的一个或多个氢原子,优选为1~5个,更优选为1~3个氢原子彼此独立地被相应数目的取代基取代。本领域技术人员能够在不付出过多努力的情况下(通过实验或理论)确定可能或不可能的取代。例如,具有游离氢的氨基或羟基与具有不饱和(如烯属)键的碳原子结合时可能是不稳定的。
[0167]“药物组合物”表示含有一种或多种本文所述化合物或其生理学上/可药用的盐或前体药物与其他化学组分的混合物,以及其他组分例如生理学/可药用的载体和赋形剂。药物组合物的目的是促进对生物体的给药,利于活性成分的吸收进而发挥生物活性。
[0168]“可药用盐”是指本公开化合物的盐,这类盐用于哺乳动物体内时具有安全性和有效性,且具有应有的生物活性。可以在化合物的最终分离和纯化过程中,或通过使合适的基团与合适的碱或酸反应来单独制备盐。通常用于形成药学上可接受的盐的碱包括无机碱,例如氢氧化钠和氢氧化钾,以及有机碱,例如氨。通常用于形成药学上可接受的盐的酸包括
无机酸以及有机酸。
[0169]
针对药物或药理学活性剂而言,术语“治疗有效量”是指无毒的但能达到预期效果的药物或药剂的足够用量。有效量的确定因人而异,取决于受体的年龄和一般情况,也取决于具体的活性物质,个案中合适的有效量可以由本领域技术人员根据常规试验确定。
[0170]
本文所用的术语“药学上可接受的”是指这些化合物、材料、组合物和/或剂型,在合理的医学判断范围内,适用于与患者组织接触而没有过度毒性、刺激性、过敏反应或其他问题或并发症,具有合理的获益/风险比,并且对预期的用途是有效。
[0171]
本文所使用的,单数形式的“一个”、“一种”和“该”包括复数引用,反之亦然,除非上下文另外明确指出。
[0172]
当将术语“约”应用于诸如ph、浓度、温度等的参数时,表明该参数可以变化
±
10%,并且有时更优选地在
±
5%之内。如本领域技术人员将理解的,当参数不是关键的时,通常仅出于说明目的给出数字,而不是限制。
[0173]
本公开的化合物还可包含其同位素衍生物。术语“同位素衍生物”指结构不同仅在于存在一种或多种同位素富集原子的化合物。例如,具有本公开的结构,除了用“氘”或“氚”代替氢,或者用
18
f
‑
氟标记(
18
f同位素)代替氟,或者用
11
c
‑
,
13
c
‑
,或者
14
c
‑
富集的碳(
11
c
‑
,
13
c
‑
,或者
14
c
‑
碳标记;
11
c
‑
,
13
c
‑
,或者
14
c
‑
同位素)代替碳原子的化合物处于本公开的范围内。这样的化合物可用作例如生物学测定中的分析工具或探针,或者可以用作疾病的体内诊断成像示踪剂,或者作为药效学、药动学或受体研究的示踪剂。
[0174]
本公开化合物的合成方法
[0175]
为了完成本公开的目的,本公开采用如下技术方案:
[0176]
方案一
[0177]
本公开通式(iiaa)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐的制备方法,包括以下步骤:
[0178][0179]
通式(iiaaa)的化合物或其盐在酸性条件下,脱去氨基保护基,得到通式(iiaa)的化合物,
[0180]
其中:
[0181]
r
w
为氨基保护基;优选为sem;
[0182]
r0‑
r4、n和q如通式(iiaa)中所定义。
[0183]
方案二
[0184]
本公开通式(iicc)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐的制备方法,包括以下步骤:
[0185][0186]
通式(iiacc)的化合物或其盐在酸性条件下,脱去氨基保护基,得到通式(iicc)的化合物,
[0187]
其中:
[0188]
r
w
为氨基保护基;优选为sem;
[0189]
r0‑
r4、n和q如通式(iicc)中所定义。
[0190]
方案三
[0191]
本公开通式(iiiaa)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐的制备方法,包括以下步骤:
[0192][0193]
通式(iiiaaa)的化合物或其盐在酸性条件下,脱去氨基保护基,得到通式(iiiaa)的化合物,
[0194]
其中:
[0195]
环中的表示单键或双键;
[0196]
r
w
、r
m
为氨基保护基;分别优选为sem、boc;
[0197]
r0‑
r4、r6、r9、n和q如通式(iiiaa)中所定义。
[0198]
方案四
[0199]
本公开通式(iiibb)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐的制备方法,包括以下步骤:
[0200][0201]
通式(iiiabb)的化合物或其盐在碱性条件下,脱去羟基保护基,得到通式(iiibb)的化合物,
[0202]
其中:
[0203]
r
n
为羟基保护基,优选为对硝基苯甲酰基;
[0204]
r0‑
r4、n和q如通式(iiibb)中所定义。
[0205]
在本公开的一些实施方案中,所述的通式(iiaaa)、(iiacc)或(iiiaaa)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中q为0、1或2。
[0206]
在本公开的一些实施方案中,所述的通式(iiaa)、(iicc)或(iiiaa)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中q为0、1、2或3;优选为0、1或2。
[0207]
在本公开的一些实施方案中,所述的通式(iiibb)或(iiiabb)所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体、或其混合物形式或其可药用的盐,其中q为0、1、2、3、4或5。
[0208]
以上合成方案中提供酸性的条件的试剂包括但不限于三氟乙酸、盐酸、氯化氢的1,4
‑
二氧六环溶液、甲酸、乙酸、硫酸、甲磺酸、硝酸、磷酸、对苯甲磺酸、me3sicl和tmsotf;优选为三氟乙酸。
[0209]
提供碱性条件的试剂包括有机碱和无机碱类,所述的有机碱类包括但不限于三乙胺、n,n
‑
二异丙基乙胺、正丁基锂、二异丙基氨基锂、醋酸钾、叔丁醇钠或叔丁醇钾,所述的无机碱类包括但不限于氢化钠、磷酸钾、碳酸钠、醋酸钠、醋酸钾、碳酸钾或碳酸铯、氢氧化钠、氢氧化锂和氢氧化钾;优选氢氧化锂。
[0210]
上述反应优选在溶剂中进行,所用溶剂包括但不限于:乙二醇二甲醚、醋酸、甲醇、乙醇、正丁醇、甲苯、四氢呋喃、二氯甲烷、石油醚、乙酸乙酯、正己烷、二甲基亚砜、1,4
‑
二氧六环、水、n,n
‑
二甲基甲酰胺及其混合物。
具体实施方式
[0211]
以下结合实施例用于进一步描述本公开,但这些实施例并非限制着本公开的范围。
[0212]
实施例
[0213]
化合物的结构是通过核磁共振(nmr)或/和质谱(ms)来确定的。nmr位移(δ)以10
‑6(ppm)的单位给出。nmr的测定是用bruker avance
‑
400核磁仪,测定溶剂为氘代二甲基亚砜
(dmso
‑
d6)、氘代氯仿(cdcl3)、氘代甲醇(cd3od),内标为四甲基硅烷(tms)。
[0214]
ms的测定用agilent 1200/1290dad
‑
6110/6120quadrupole ms液质联用仪(生产商:agilent,ms型号:6110/6120quadrupole ms)、waters acquity uplc
‑
qd/sqd(生产商:waters,ms型号:waters acquity qda detector/waters sq detector)、thermo ultimate 3000
‑
q exactive(生产商:thermo,ms型号:thermo q exactive)。
[0215]
高效液相色谱法(hplc)分析使用agilent hplc 1200dad、agilent hplc1200vwd和waters hplc e2695
‑
2489高压液相色谱仪。
[0216]
手性hplc分析测定使用agilent 1260dad高效液相色谱仪。
[0217]
高效液相制备使用waters 2545
‑
2767、waters 2767
‑
sq detecor2、shimadzu lc
‑
20ap和gilson gx
‑
281制备型色谱仪。
[0218]
手性制备使用shimadzu lc
‑
20ap制备型色谱仪。
[0219]
combiflash快速制备仪使用combiflash rf200(teledyne isco)。
[0220]
薄层层析硅胶板使用烟台黄海hsgf254或青岛gf254硅胶板,薄层色谱法(tlc)使用的硅胶板采用的规格是0.15mm~0.2mm,薄层层析分离纯化产品采用的规格是0.4mm~0.5mm。
[0221]
硅胶柱色谱法一般使用烟台黄海硅胶200~300目硅胶为载体。
[0222]
激酶平均抑制率及ic
50
值的测定用novostar酶标仪(德国bmg公司)。
[0223]
本公开的已知的起始原料可以采用或按照本领域已知的方法来合成,或可购买自abcr gmbh&co.kg,acros organics,aldrich chemical company,韶远化学科技(accela chembio inc)、达瑞化学品等公司。
[0224]
实施例中无特殊说明,反应能够均在氩气氛或氮气氛下进行。
[0225]
氩气氛或氮气氛是指反应瓶连接一个约1l容积的氩气或氮气气球。
[0226]
氢气氛是指反应瓶连接一个约1l容积的氢气气球。
[0227]
加压氢化反应使用parr 3916ekx型氢化仪和清蓝ql
‑
500型氢气发生器或hc2
‑
ss型氢化仪。
[0228]
氢化反应通常抽真空,充入氢气,反复操作3次。
[0229]
微波反应使用cem discover
‑
s 908860型微波反应器。
[0230]
实施例中无特殊说明,溶液是指水溶液。
[0231]
实施例中无特殊说明,反应的温度为室温,为20℃~30℃。
[0232]
实施例中的反应进程的监测采用薄层色谱法(tlc),反应所使用的展开剂,纯化化合物采用的柱层析的洗脱剂的体系和薄层色谱法的展开剂体系包括:a:二氯甲烷/甲醇体系,b:正己烷/乙酸乙酯体系,c:石油醚/乙酸乙酯体系,溶剂的体积比根据化合物的极性不同而进行调节,也可以加入少量的三乙胺和醋酸等碱性或酸性试剂进行调节。
[0233]
其中boc为叔丁氧羰基;sem为(三甲基硅基)乙氧基甲基。
[0234]
实施例1
[0235]
(s)
‑4‑
(5
‑
(2
‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮1
[0236][0237][0238]
第一步
[0239]4‑
氯
‑
5,5
‑
二甲基
‑7‑
((2
‑
(三甲基硅)乙氧基)甲基)
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮1b
[0240]4‑
氯
‑
5,5
‑
二甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮1a(2.3g,11.64mmol,上海毕得科技医药有限公司)室温下溶解于n,n
‑
二甲基甲酰胺(40ml)中,加入氢化钠(930mg,23.25mmol,60%purity),搅拌30分钟,加入2
‑
(氯甲氧基)乙基三甲基硅烷(2.91g,17.45mmol),室温搅拌2小时。反应液倒入30ml水中,用乙酸乙酯(30ml*2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物1b(2.5g),产率66%。
[0241]
ms m/z(esi):328.1[m+1]
+
[0242]
第二步
[0243]2‑
(5,5
‑
二甲基
‑6‑
氧代
‑7‑
((2
‑
(三甲硅基)乙氧基)甲基)
‑
6,7
‑
二氢
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
4,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(2h)
‑
羧酸叔丁酯1d
[0244]
化合物1b(2.5g,7.6mmol)室温溶解于n,n
‑
二甲基甲酰胺(20ml)中,加入4,6
‑
二氢吡咯[3,4
‑
c]吡唑
‑
5(2h)
‑
羧酸叔丁酯1c(1.59g,7.6mmol,南京药石科技股份有限公司)和
碳酸铯(7.45g,22.87mmol),然后微波140℃,搅拌1小时。反应液倒入30ml水中,用乙酸乙酯(30ml*2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物1d(1.7g),产率44%。
[0245]
ms m/z(esi):501.1[m+1]
+
[0246]
第三步
[0247]4‑
(5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑7‑
((2
‑
(三甲基硅基)乙氧基)甲基)
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮1e
[0248]
化合物1d(1.7g,3.40mmol)溶解于4m盐酸二氧六环(15ml)。反应搅拌过夜,减压浓缩得到标题化合物1e(1.36g),产率99%,不经纯化直接做下一步反应。
[0249]
ms m/z(esi):401.9[m+1]
+
[0250]
第四步
[0251]
(s)
‑
(2
‑
(4
‑
氯苯基)
‑3‑
(2
‑
(5,5
‑
二甲基
‑6‑
氧代
‑7‑
((2
‑
(三甲基硅基)乙氧基)甲基)
‑
6,7
‑
二氢
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)吡咯并[3,4
‑
c]吡唑
‑
5(2h,4h,6h)
‑
基)
‑3‑
氧代丙基(异丙基)氨基甲酸叔丁酯1g
[0252]
化合物1e(1.36g,3.40mmol)室温下溶解于n,n
‑
二甲基甲酰胺(5ml),然后加入(s)
‑3‑
((叔丁氧羰基)(异丙基)氨基)
‑2‑
(4
‑
氯苯基)丙酸1f(1.27g,3.71mmol,按照org.process res.dev.2014,18,12,1652
‑
1666公开的方法合成)和2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(1.65g,5.08mmol,韶远化学科技(上海)有限公司),二异丙基乙基胺(1.31g,10.15mmol),室温反应20小时。反应液倒入30ml水中,用乙酸乙酯(30ml*2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物1g(2.2g),产率89%。ms m/z(esi):724.1[m+1]
+
[0253]
第五步
[0254]
(s)
‑4‑
(5
‑
(2
‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮1
[0255]
化合物1g(3.5g,4.83mmol)在冰浴下溶解于二氯甲烷(20ml)中,加入三氟乙酸(5ml),室温搅拌2小时。反应液减压浓缩,加入甲醇(40ml)、水(8ml)和碳酸钾(3.33g,24.1mmol),搅拌过夜,反应液加水淬灭,二氯甲烷萃取(30ml
×
3),有机相用无水硫酸钠干燥,减压浓缩后通过反相制备纯化(仪器型号:gilson 281色谱柱:sharpsil
‑
t,prep30*150mm;5μm;c18流动相:a
‑
水(0.1%三氟乙酸)b
‑
乙腈流速:30ml/min柱温:室温)得到目标化合物1(1.8g),产率61%。
[0256]
ms m/z(esi):494.1[m+1]
+
[0257]1h nmr(400mhz,cd3od):δ8.61
‑
8.52(m,2h),7.55
‑
7.38(m,4h),4.90(dd,1h),4.79(d,1h),4.65(dd,1h),4.37
‑
4.31(m,1h),4.24(t,1h),3.80
‑
3.70(m,1h),3.54
‑
3.43(m,1h),2.52(dd,1h),1.61(s,3h),1.60(dd,3h),1.39(t,6h)。
[0258]
实施例2
[0259]
(s)
‑4‑
(5
‑
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑5‑
甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮2
‑
p1
[0260][0261]
(r)
‑4‑
(5
‑
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑5‑
甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮2
‑
p2
[0262][0263]
第一步
[0264]4‑
氯
‑5‑
甲基
‑6‑
((2
‑
(三甲硅基)乙氧基)甲氧基)
‑7‑
((2
‑
(三甲硅基)乙氧基)甲基)
‑
7h吡咯并[2,3
‑
d]嘧啶2b
[0265]4‑
氯
‑5‑
甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮2a(120mg,653.61μmol,上海韶远科技有限公司)溶于n,n
‑
二甲基甲酰胺(2ml),冰水浴下加入nah(52mg,1.3mmol,60%purity)反应30分钟,加入semcl(218mg,1.3076mmol,231.4225μl),室温反应过夜。冰水浴下氯化铵溶液淬灭,乙酸乙酯稀释,水洗后有机相干燥浓缩,用柱层析以洗脱剂体系c纯化得到标题化合物2b(144mg),产率50%。ms m/z(esi):444.1[m+1]
+
[0266]
第二步
[0267]2‑
(5
‑
甲基
‑6‑
((2
‑
(三甲硅基)乙氧基)甲氧基)
‑7‑
((2
‑
(三甲硅基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
4,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(2h)
‑
羧酸叔丁酯2d
[0268]
化合物2b(144mg,324.24μmol)和化合物1c(135mg,645.18μmol)溶于n,n
‑
二甲基甲酰胺(1.5ml)加入碳酸铯(317mg,972.93μmol)混合,微波140℃反应1小时。乙酸乙酯稀释,有机相水洗后,干燥浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物2d(70mg),产率35%。
[0269]
ms m/z(esi):617.3[m+1]
+
[0270]
第三步
[0271]4‑
(5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑5‑
甲基
‑7‑
((2
‑
(三甲硅基)乙氧基)甲基)
‑
5h吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮2e
[0272]
化合物2d(20mg,41.09μmol)溶解于4m盐酸二氧六环(2ml)中,室温搅拌2小时,反应液减压浓缩得到粗品2e(10mg),产率94%,未纯化直接进行下一步反应。
[0273]
ms m/z(esi):387.1[m+1]
+
[0274]
第四步
[0275]
((2s)
‑2‑
(4
‑
氯苯基)
‑3‑
(2
‑
(5
‑
甲基
‑6‑
氧代
‑7‑
((2
‑
(三甲基硅基)乙氧基)甲基)
‑
6,7
‑
二氢
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)吡咯并[3,4
‑
c]吡唑
‑
5(2h,4h,6h)
‑
基)
‑3‑
氧代丙基(异丙基)氨基甲酸叔丁酯2g
[0276]
化合物2e(10mg,25.87μmol)室温下溶解于n,n
‑
二甲基甲酰胺(2ml)中,加入化合物1f(9mg,26.32μmol),二异丙基乙基胺(10mg,77.52μmol)和2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(12mg,36.92μmol,韶远化学科技(上海)有限公司),室温搅拌过夜。反应液倒入10ml水中,用乙酸乙酯(20ml*2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩得到粗品2g(15mg),产率:82%,未纯化直接进行下一步反应。
[0277]
ms m/z(esi):711.3[m+1]
+
[0278]
第五步
[0279]
(s)
‑4‑
(5
‑
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑5‑
甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮2
‑
p1
[0280]
(r)
‑4‑
(5
‑
((s)
‑2‑
(4
‑
chlorophenyl)
‑3‑
(isopropylamino)propanoyl)
‑
5,6
‑
dihydropyrrolo[3,4
‑
c]pyrazol
‑
2(4h)
‑
yl)
‑5‑
methyl
‑
5h
‑
pyrrolo[2,3
‑
d]pyrimidin
‑
6(7h)
‑
one
[0281]
(r)
‑4‑
(5
‑
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑5‑
甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮2
‑
p2
[0282]
化合物2g(30mg,42.23μmol)溶解于二氯甲烷(4ml),加入三氟乙酸(1ml),室温搅拌2小时。反应液减压浓缩,加入甲醇(5ml)、水(1ml)和碳酸钾(29mg,0.21mmol),搅拌过夜,反应液加水淬灭,二氯甲烷萃取(10ml
×
3),有机相用无水硫酸钠干燥,减压浓缩后通过反相制备纯化(仪器型号:gilson 281色谱柱:sharpsil
‑
t,prep30*150mm;5μm;c18流动相:a
‑
水(0.1%三氟乙酸)b
‑
乙腈流速:30ml/min柱温:室温)得到目标化合物(5mg,保留时间15.01min,产率20%)和(5mg,保留时间13.72min,产率20%)。
[0283]
单一构型化合物(保留时间:15.01min):
[0284]
ms m/z(esi):480.1[m+1]
+
[0285]1h nmr(400mhz,cd3od):δ8.65
‑
8.43(m,2h),7.61
‑
7.27(m,4h),4.85
‑
4.53(m,2h),4.44
‑
4.16(m,2h),3.97
‑
3.80(m,1h),3.80
‑
3.67(m,1h),3.54
‑
3.40(m,1h),3.30
‑
3.16(m,1h),2.96(d,1h),1.64
‑
1.45(m,3h),1.39(s,3h),1.35(d,3h)。
[0286]
单一构型化合物(保留时间:13.72min):
[0287]
ms m/z(esi):480.0[m+1]
+
[0288]1h nmr(400mhz,cd3od):δ8.55(s,1h),8.51(d,1h),7.51
‑
7.28(m,4h),5.10
‑
4.67(m,2h),4.67
‑
4.50(m,1h),4.46
‑
4.30(m,1h),4.21
‑
4.02(m,1h),3.51
‑
3.25(m,2h),3.00
‑
2.77(m,2h),1.3(s,3h),1.15(d,3h),1.12(d,3h)。
[0289]
实施例3
[0290]
(s)
‑2‑
(4
‑
氯苯基)
‑1‑
(2
‑
((5r,7r)
‑7‑
羟基
‑5‑
甲基
‑
6,7
‑
二氢
‑
5h
‑
环戊并[d]嘧啶
‑4‑
基)吡咯并[3,4
‑
c]吡唑
‑
5(2h,4h,6h)
‑
基)
‑3‑
(异丙基氨基)丙
‑1‑
酮3
[0291][0292]
[0293]
第一步
[0294]2‑
((5r,7r)
‑5‑
甲基
‑7‑
((4
‑
硝基苯甲酰基)氧基)
‑
6,7
‑
二氢
‑
5h
‑
环戊并[d]嘧啶
‑4‑
基)
‑
4,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(2h)
‑
羧酸叔丁酯3b
[0295]
化合物1c(60mg,286.75μmol)室温溶解于n,n
‑
二甲基甲酰胺(5ml),然后加入氢化钠(13.76mg,344.10μmol,60%purity)室温搅拌1小时。然后加入化合物3a(76.56mg,229.40μmol,采用专利申请“cn104876921a”中说明书第51页的公开方法制备得到)的n,n
‑
二甲基甲酰胺(0.5ml)溶液,室温搅拌1小时。用柱层析以洗脱剂体系c纯化得到标题化合物3b(10mg),产率10%。
[0296]
ms m/z(esi):507.1[m+1]
+
[0297]
第二步
[0298]
(5r,7r)
‑4‑
(5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑5‑
甲基
‑
6,7
‑
二氢
‑
5h
‑
环戊并[d]嘧啶
‑7‑
基4
‑
硝基苯甲酸酯3c
[0299]
化合物3b(15mg,29.61μmol)加入4m盐酸二氧六环溶液(2ml),室温搅拌2小时。减压浓缩后得到粗品3c(12mg),产率99%。未经纯化直接进行下一步反应。
[0300]
ms m/z(esi):407.1[m+1]
+
[0301]
第三步(5r,7r)
‑4‑
(5
‑
((s)
‑3‑
((叔丁氧羰基)(异丙基)氨基)
‑2‑
(4
‑
氯苯基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑5‑
甲基
‑
6,7
‑
二氢
‑
5h
‑
环戊并[d]嘧啶
‑7‑
基4
‑
硝基苯甲酸酯3d
[0302]
化合物3c(12mg,29.53μmol)室温溶于n,n
‑
二甲基甲酰胺(2ml),然后加入化合物1f(10.09mg,29.53μmol),2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(13.46mg,35.43μmol,韶远化学科技(上海)有限公司),室温搅拌2小时。反应液倒入10ml水中,用乙酸乙酯(20ml*2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩得到粗品3d(20mg),产率:93%,未纯化直接进行下一步反应。
[0303]
ms m/z(esi):730.4[m+1]
+
[0304]
第四步(5r,7r)
‑4‑
(5
‑
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑5‑
甲基
‑
6,7
‑
二氢
‑
5h
‑
环戊并[d]嘧啶
‑7‑
基4
‑
硝基苯甲酸酯3e
[0305]
化合物3d(20mg,27.39μmol)加入4m盐酸二氧六环溶液(2ml),室温搅拌1小时。减压浓缩后得到粗品3e(17mg),产率100%,未纯化直接进行下一步反应。ms m/z(esi):631.8[m+1]
+
[0306]
第五步
[0307]
(s)
‑2‑
(4
‑
氯苯基)
‑1‑
(2
‑
((5r,7r)
‑7‑
羟基
‑5‑
甲基
‑
6,7
‑
二氢
‑
5h
‑
环戊并[d]嘧啶
‑4‑
基)吡咯并[3,4
‑
c]吡唑
‑
5(2h,4h,6h)
‑
基)
‑3‑
(异丙基氨基)丙
‑1‑
酮3
[0308]
化合物3e(20mg,31.74μmol)溶解于水(2ml)和四氢呋喃溶液(3ml),然后加入氢氧化锂(2.66mg,111.09μmol)室温搅拌1小时。减压浓缩,残余物用液相制备纯化(仪器型号:gilson 281色谱柱:x
‑
bridge,prep 30*150mm;5μm;c18流动相:a
‑
水(10mm碳酸氢铵)b
‑
乙腈流速:30ml/min柱温:室温)得到标题化合物3(2mg),产率:13%
[0309]
ms m/z(esi):481.9[m+1]
+
[0310]
1h nmr(400mhz,cdcl3):δ8.73(s,1h),8.40(s,1h),7.41
‑
7.30(m,4h),5.48
‑
5.23
(m,1h),4.87
‑
4.54(m,2h),4.47
‑
4.19(m,2h),4.17
‑
3.92(m,2h),3.91
‑
3.79(m,1h),3.67
‑
3.50(m,1h),3.19
‑
2.98(m,1h),2.98
‑
2.73(m,2h),2.53
‑
2.35(m,1h),2.33
‑
2.15(m,1h),1.40
‑
1.26(m,6h),1.17(s,3h)。
[0311]
实施例4
[0312]
(s)
‑1‑
(2
‑
(7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)吡咯并[3,4
‑
c]吡唑
‑
5(2h,4h,6h)
‑
基)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙
‑1‑
酮4
[0313][0314]
第一步
[0315]2‑
(7
‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
4,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(2h)
‑
羧酸叔丁酯4b
[0316]
化合物1c(3.00g,14.33mmol,南京药石科技股份有限公司)溶于n,n
‑
二甲基甲酰胺(50ml),降温至0℃,加入氢化钠(0.630g,15.75mmol,60%),搅拌反应60分钟,加入4
‑
氯
‑7‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶4a(4.07g,14.33mmol,采用专利申请“ep3360878a1”中说明书第33页的公开方法制备得到),室温搅拌14小时。冰水浴下,加入饱和氯化铵溶液,乙酸乙酯萃取,有机相无水硫酸钠干燥后减压浓缩,用柱层析以洗脱剂体系b纯化得到标题化合物4b(3.5g),产率:53%。
[0317]
ms m/z(esi):457.0[m+1]
+
[0318]
第二步
[0319]4‑
(5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑7‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶4c
[0320]
将化合物4b(0.600g,1.31mmol)溶于甲醇(10ml),加入盐酸二氧六环溶液(3ml,4m),反应17小时。浓缩反应液,经纯化得到粗品标题化合物4c(320mg),产率:68%。
[0321]
ms m/z(esi):357.8[m+1]
+
[0322]
第三步
[0323]
(s)
‑
(2
‑
(4
‑
氯苯基)
‑3‑
氧代
‑3‑
(2
‑
(7
‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)吡咯并[3,4
‑
c]吡唑
‑
5(2h,4h,6h)
‑
基)丙基)(异丙基)氨基甲酸叔丁酯4d
[0324]
将化合物4c(0.120g,0.33mmol),化合物1f(115mg,0.33mmol)溶于干燥的n,n
‑
二甲基甲酰胺(5ml)中,冰浴下加入2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(127mg,0.33mmol,韶远化学科技(上海)有限公司),二异丙基乙基胺(0.130g,0.99mmol),氮气保护,室温搅拌过夜,加水淬灭,乙酸乙酯萃取,无水硫酸钠干燥,减压浓缩后,得到标题化合物4d(200mg),产率:88%。
[0325]
ms m/z(esi):682.2[m+1]
+
[0326]
第四步
[0327]
(s)
‑1‑
(2
‑
(7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)吡咯并[3,4
‑
c]吡唑
‑
5(2h,4h,6h)
‑
基)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙
‑1‑
酮4
[0328]
将化合物4d(200mg,0.29mmol)溶于干燥的二氯甲烷(5ml)中,加入三氟乙酸(2ml),室温反应17小时。减压浓缩干,加入四氢呋喃(10ml),水(2ml)中,加入碳酸钾(0.405g,2.93mmol)搅拌5小时,反应液加水淬灭,乙酸乙酯萃取(20ml
×
3),有机相无水硫酸钠干燥,减压浓缩,粗品制备hplc(色谱柱:x
‑
bridge,prep 30*150mm;5μm;流动相:a
‑
水(10mm碳酸氢铵)b
‑
乙腈,梯度配比:a20%
‑
45%),得到标题化合物4(20mg),产率3%。
[0329]
ms m/z(esi):450.1[m+1]
+
[0330]1h nmr(400mhz,d6
‑
dmso)δ8.63(s,1h),8.61(s,1h),7.58(d,1h),7.50
‑
7.35(m,4h),7.05(dd,1h),5.06(d,1h),4.95(d,1h),4.74
‑
4.29(m,4h),4.13
‑
4.04(m,1h),3.25
‑
3.11(m,1h),2.85
‑
2.65(m,2h),0.97(t,6h)。
[0331]
实施例5
[0332]1‑
(2
‑
(7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)
‑3‑
氨基
‑2‑
(4
‑
氯苯基)丙
‑1‑
酮5
[0333][0334]
第一步(2
‑
(4
‑
氯苯基)
‑3‑
氧代
‑3‑
(2
‑
(7
‑
((2
‑
(三甲基硅烷基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)丙基)氨基甲酸叔丁酯5b
[0335]
将化合物4
‑
(5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑7‑
((2
‑
(三甲基硅烷基)乙氧基)甲基
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶4c(50mg,0.14mmol)溶于n,n
‑
二甲基甲酰胺(2ml)中,加入3
‑
((叔丁氧羰基)氨基)
‑2‑
(4
‑
氯苯基)丙酸5a(42mg,0.14mmol,按照wo2005051304公开的方法合成),2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(64mg,0.17mmol),二异丙基乙基胺(54mg,0.42mmol),室温反应2小时。加入乙酸乙酯(20ml)稀释,饱和碳酸氢钠溶液洗涤。有机相干燥后减压浓缩,得到标题化合物5b(60mg),产物不经纯化,直接用于下一步反应。
[0336]
ms m/z(esi):638.2[m+1]
+
[0337]
第二步
[0338]1‑
(2
‑
(7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)
‑3‑
氨基
‑2‑
(4
‑
氯苯基)丙
‑1‑
酮5
[0339]
将化合物5b(35mg,0.55mmol)溶于二氯甲烷(4ml)和三氟乙酸(4ml)中,室温搅拌反应1小时。反应液减压浓缩后加入四氢呋喃(2ml)稀释,加入饱和碳酸钾溶液(2ml),室温搅拌5小时,乙酸乙酯萃取(10ml
×
5),无水硫酸钠干燥后,过滤减压浓缩,残余物用液相制备纯化(仪器型号:gilson 281色谱柱:x
‑
bridge,prep30
×
150mm;5μm;c18流动相:a
‑
水(10mm碳酸氢铵)b
‑
乙腈流速:30ml/min柱温:室温)得到标题化合物5(10mg),产率:4%。
[0340]
ms m/z(esi):408.1[m+1]
+
[0341]1h nmr(400mhz,cd3od)δ8.43
‑
8.51(m,2h),7.31
‑
7.33(m,4h),7.06
‑
7.11(m,1h),4.86
‑
4.89(m,1h),4.48
‑
4.52(m,2h),4.28
‑
4.22(m,1h),3.97
‑
3.98(m,1h),3.48
‑
3.52(m,1h),3.24
‑
3.25(m,1h),2.88
‑
2.90(m,1h)。
[0342]
实施例6
[0343]
(s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)
‑1‑
(2
‑
(5
‑
甲基
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)丙
‑1‑
酮6
[0344][0345]
第一步
[0346]4‑
氯
‑5‑
甲基
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶6b
[0347]
氮气保护下,5
‑
溴
‑4‑
氯
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶6a(2.0g,8.60mmol,上海毕得科技医药有限公司)溶解于四氢呋喃(30ml)中,
‑
78℃下缓慢滴加正丁基锂(7.74ml,19.3mmol,2.5m的四氢呋喃溶液,安耐吉试剂有限公司),并在该温度下搅拌30分钟。滴加碘甲烷(1.95g,13.7mmol,阿达玛斯试剂有限公司),室温搅拌2小时。反应液倒入饱和氯化铵溶液中,用乙酸乙酯(30ml
×
2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物6b(700mg),产率48%。
[0348]
ms m/z(esi):168.3[m+1]
+
[0349]
第二步
[0350]4‑
氯
‑5‑
甲基
‑7‑
(2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶6c
[0351]
将化合物6b(500mg,2.98mmol)室温溶解于n,n
‑
二甲基甲酰胺二甲基(5ml)中,冰水浴下加入氢化钠(143mg,5.95mmol,纯度60%),搅拌30分钟后加入2
‑
(三甲基硅烷基)乙氧甲基氯(746mg,4.47mmol),室温搅拌2小时。反应液倒入50ml冰水中,用乙酸乙酯(50ml
×
2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物6c(700mg),产率78%。
[0352]
ms m/z(esi):298.1[m+1]
+
[0353]
第三步
[0354]2‑
(5
‑
甲基
‑7‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
羧酸叔丁酯6d
[0355]
将化合物1c(210mg,1.00mmol)溶解于n,n
‑
二甲基甲酰胺二甲基(3ml)中,冰水浴下加入氢化钠(26mg,0.68mmol,纯度60%),搅拌1小时后加入6c(200mg,0.67mmol),室温搅拌2小时。反应液倒入30ml冰水中,用乙酸乙酯(30ml
×
2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤后减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物6d(200mg),产率63%。
[0356]
ms m/z(esi):471.2[m+1]
+
[0357]
第四步
[0358]4‑
(5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑5‑
甲基
‑7‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶6e
[0359]
将化合物6d(200mg,0.42mmol)溶解于4m盐酸二氧六环溶液(5ml)中,室温搅拌2小时。减压浓缩后得到标题化合物6e(150mg),产物不经纯化,直接用于下一步反应。
[0360]
ms m/z(esi):371.7[m+1]
+
[0361]
第五步
[0362]
(s)
‑
(2
‑
(4
‑
氯苯基)
‑3‑
(2
‑
(5
‑
甲基
‑7‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)
‑3‑
氧代丙基(异丙基)氨基甲酸叔丁酯6f
[0363]
将化合物6e(100mg,0.27mmol)溶解于n,n
‑
二甲基甲酰胺(5ml)中,加入(s)
‑3‑
((叔丁氧羰基)(异丙基)氨基)
‑2‑
(4
‑
氯苯基)丙酸1f(92mg,0.27mmol)和2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(153mg,0.40mmol),二异丙基乙基胺(104mg,0.81mmol),室温反应20小时。反应液倒入水(30ml)中,用乙酸乙酯(30ml
×
2)萃取,有机相用饱和氯化钠溶液洗涤,减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物6f(130mg),产率69%。
[0364]
ms m/z(esi):694.3[m+1]
+
[0365]
第六步
[0366]
(s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)
‑1‑
(2
‑
(5
‑
甲基
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)丙
‑1‑
酮6
[0367]
将化合物6f(100mg,0.14mmol)溶解于二氯甲烷(5ml)中,加入三氟乙酸(2ml),室温搅拌2小时。反应液减压浓缩,加入甲醇,水,碳酸钾调节ph值至碱性,搅拌过夜。反应液加水淬灭,二氯甲烷萃取(30ml
×
2),有机相无水硫酸钠干燥,减压浓缩后残余物用液相制备纯化(仪器型号:gilson 281色谱柱:x
‑
bridge,prep 30
×
150mm;5μm;c18流动相:a
‑
水(10mm碳酸氢铵)b
‑
乙腈流速:30ml/min柱温:室温)得到标题化合物6(10mg),产率15%。
[0368]
ms m/z(esi):464.2[m+1]
+
[0369]1h nmr(400mhz,cd3od):δ8.49(s,1h),8.33
‑
8.37(m,1h),7.40
‑
7.41(m,4h),7.22(s,1h),4.71
‑
4.75(m,1h),4.55
‑
4.61(m,1h),4.33
‑
4.37(m,1h),4.11
‑
4.13(m,1h),3.38
‑
3.40(m,2h),2.82
‑
2.95(m,2h),2.37
‑
2.39(m,3h),1.11
‑
1.15(m,6h)。
[0370]
实施例7
[0371]
(s)
‑2‑
(4
‑
氯苯基)
‑1‑
((s)
‑4‑
(羟甲基)
‑2‑
(7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)
‑3‑
(异丙基氨基)丙
‑1‑
酮7
[0372][0373][0374]
第一步
[0375]5‑
(叔丁基)4
‑
甲基(s)
‑2‑
(7
‑
((2
‑
(三甲基硅基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
4,5(4h)
‑
二羧酸酯7b
[0376]
将化合物5
‑
(叔丁基)4
‑
甲基(s)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
4,5(4h)
‑
二羧酸酯7a(410mg,1.53mmol,按照专利tw2017/8221中公开的方法合成)溶于n,n
‑
二甲基甲酰胺(4ml)中,冰水浴下加入氢化钠(123mg,3.08mmol),搅拌30分钟后加入4a(435mg,1.53mmol),室温反应2小时。加入乙酸乙酯(100ml)稀释,饱和碳酸氢钠溶液洗涤。有机相干燥后减压浓缩,用柱层析以洗脱剂体系c纯化得到标题化合物7b(300mg),产率:38%。
[0377]
ms m/z(esi):515.2[m+1]
+
[0378]
第二步
[0379]
(s)
‑4‑
(羟甲基)
‑2‑
(7
‑
((2
‑
(三甲基硅基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
羧酸叔丁酯7c
[0380]
将化合物7b(300mg,0.58mmol)溶于四氢呋喃(3ml),加入硼氢化锂(38mg,1.74mmol),室温搅拌反应16小时,加入乙酸乙酯稀释,饱和碳酸氢钠溶液洗涤。有机相干燥后减压浓缩,得到标题化合物7c(280mg),产物不经纯化,直接用于下一步反应。
[0381]
ms m/z(esi):487.2[m+1]
+
[0382]
第三步
[0383]
(s)
‑
(2
‑
(7
‑
((2
‑
(三甲基硅基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,4,5,6
‑
四氢吡咯并[3,4
‑
c]吡唑
‑4‑
基)甲醇7d
[0384]
将化合物7c(150mg,0.31mmol)溶于4m的盐酸二氧六环溶液(2ml),室温反应1小时,将反应液浓缩后,得到标题化合物7d(100mg),产物不经纯化,直接用于下一步反应。
[0385]
ms m/z(esi):387.4[m+1]
+
[0386]
第四步
[0387]
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
((s)
‑4‑
(羟甲基)
‑2‑
(7
‑
((2
‑
三甲基硅基)乙氧基)甲基)
‑
7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)
‑3‑
氧丙基)(异丙基)氨基甲酸叔丁酯7e
[0388]
将化合物7d(60mg,0.16mmol)溶于n,n
‑
二甲基甲酰胺(2ml)中,加入化合物1f(53mg,0.16mmol),2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(71mg,0.19mmol),二异丙基乙基胺(60mg,0.46mmol),室温反应20小时。加入乙酸乙酯稀释,饱和碳酸氢钠溶液洗涤。有机相干燥后减压浓缩,用柱层析以洗脱剂体系c纯化得到标题化合物7e(100mg),产率:91%。
[0389]
ms m/z(esi):710.2[m+1]
+
[0390]
第五步
[0391]
(s)
‑2‑
(4
‑
氯苯基)
‑1‑
((s)
‑4‑
(羟甲基)
‑2‑
(7h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)
‑3‑
(异丙基氨基)丙
‑1‑
酮7
[0392]
将化合物7e(100mg,0.14mmol)溶于二氯甲烷(2ml)和三氟乙酸(2ml)的混合溶剂中,室温搅拌反应1小时,反应液减压浓缩。后加入四氢呋喃(2ml)稀释,加入饱和碳酸钾溶液(2ml),室温搅拌5小时,反应液乙酸乙酯萃取(10ml
×
5),有机相浓缩有机相干燥后减压浓缩,残余物用液相制备纯化(仪器型号:gilson 281色谱柱:x
‑
bridge,prep 30
×
150mm;5μm;c18流动相:a
‑
水(10mm碳酸氢铵)b
‑
乙腈流速:30ml/min柱温:室温)得到标题化合物7(15mg),产率22%。
[0393]
ms m/z(esi):480.1[m+1]
+
[0394]1h nmr(400mhz,cd3od)δ8.44
‑
8.45(m,2h),7.36
‑
7.44(m,5h),7.02
‑
7.03(m,1h),5.20
‑
5.24(m,1h),4.78
‑
4.80(m,1h),4.23
‑
4.25(m,2h),4.06
‑
4.10(m,1h),3.90
‑
3.94(m,1h),3.36
‑
3.41(m,1h),3.07
‑
3.20(m,2h),1.23
‑
1.33(m,6h)。
[0395]
实施例8
[0396]4‑
((s)
‑5‑
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基基)丙酰基)
‑4‑
(羟甲基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮8
[0397][0398]
第一步
[0399]
(s)
‑4‑
(羟甲基)
‑
4,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(2h)
‑
羧酸叔丁酯8a
[0400]
将化合物7a(50mg,0.19mmol)溶于四氢呋喃(2ml)中,冰水浴下加入硼氢化锂(20mg,0.92mmol),室温搅拌15小时。加入乙酸乙酯稀释,饱和碳酸氢钠溶液洗涤。有机相干燥后减压浓缩,用柱层析以洗脱剂体系c纯化得到标题化合物8a(30mg),产率:67%。
[0401]
ms m/z(esi):240.1[m+1]
+
[0402]
第二步
[0403]
(s)
‑2‑
(5,5
‑
二甲基
‑6‑
氧代
‑7‑
((2
‑
(三甲基硅基)乙氧基)甲基)
‑
6,7
‑
二氢
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑4‑
(羟甲基)
‑
4,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(2h)
‑
羧酸叔丁酯8b
[0404]
将化合物8a(30mg,0.13mmol)溶于n,n
‑
二甲基甲酰胺(2ml)中,冰水浴下加入氢化钠(30mg,0.75mmol)。室温搅拌30分钟,加入1a(41mg,0.13mmol),室温反应2小时。加入乙酸乙酯(50ml)稀释,饱和碳酸氢钠溶液洗涤。有机相干燥后过滤减压浓缩,用柱层析以洗脱剂体系c纯化得到标题化合物8b(33mg),产率:50%。
[0405]
ms m/z(esi):531.2[m+1]
+
[0406]
第三步
[0407]
(s)
‑4‑
(4
‑
(羟甲基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑7‑
((2
‑
(三甲基硅基)乙氧基)甲基)
‑
5h吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮8c
[0408]
将化合物8b(35mg,0.66mmol)溶于4m的盐酸二氧六环溶液(2ml),室温反应1小时,将反应液浓缩后,得到标题化合物8c(28mg),产物不经纯化,直接用于下一步反应。
[0409]
ms m/z(esi):431.2[m+1]
+
[0410]
第四步
[0411]
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
((s)
‑2‑
(5,5
‑
二甲基
‑6‑
氧代
‑7‑
((2
‑
(三甲基硅基)乙氧基)甲基)
‑
6,7
‑
二氢
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑4‑
(羟甲基)吡咯并[3,4
‑
c]吡唑
‑
5(2h,4h,6h)
‑
基)
‑3‑
氧丙基)(异丙基)氨基甲酸叔丁酯8d
[0412]
将化合物8c(30mg,0.07mmol)溶于n,n
‑
二甲基甲酰胺(2ml)中,加入1f(24mg,0.07mmol),2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(40mg,0.11mmol),二异丙基乙基胺(32mg,0.25mmol),室温反应20小时。加入乙酸乙酯稀释,饱和碳酸氢钠溶液洗涤。有机相干燥后减压浓缩,用柱层析以洗脱剂体系c纯化得到标题化合物8d(30mg),产率:57%。
[0413]
ms m/z(esi):754.1[m+1]
+
[0414]
第五步
[0415]4‑
((s)
‑5‑
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑4‑
(羟甲基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑
6(7h)
‑
酮8
[0416]
将化合物8d(30mg,0.04mmol)溶于二氯甲烷(2ml)和三氟乙酸(2ml)的混合溶剂中,室温搅拌反应1小时。反应液减压浓缩后加入四氢呋喃(2ml)稀释,加入饱和碳酸钾溶液(2ml),室温搅拌5小时,乙酸乙酯萃取(10ml
×
5),有机相浓缩有机相干燥后减压浓缩,残余物用液相制备纯化(仪器型号:gilson 281色谱柱:x
‑
bridge,prep 30
×
150mm;5μm;c18流动相:a
‑
水(10mm碳酸氢铵)b
‑
乙腈流速:30ml/min柱温:室温)得到标题化合物8(15mg),产率71%。
[0417]
ms m/z(esi):524.2[m+1]
+
[0418]1h nmr(400mhz,cd3od)δ8.61(s,1h),8.57(s,1h),7.41
‑
7.47(m,4h),5.22
‑
5.24(m,1h),4.93
‑
4.97(m,1h),4.44
‑
4.50(m,1h),4.09
‑
4.13(m,2h),3.93
‑
3.96(m,1h),3.36
‑
3.42(m,1h),2.86
‑
3.03(m,2h),1.54
‑
1.62(m,6h),1.12
‑
1.20(m,6h)。
[0419]
实施例9
[0420]4‑
((r)
‑5‑
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑4‑
甲基
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5,7
‑
二氢
‑
6h
‑
吡咯并[2,3
‑
d]嘧啶
‑6‑
酮9
[0421][0422]
第一步
[0423]
(2r)
‑2‑
甲基
‑4‑
氧代
‑
吡咯烷
‑1‑
羧酸叔丁酯9b
[0424]
将(2r,4r)
‑4‑
羟基
‑2‑
甲基
‑
吡咯烷
‑1‑
羧酸叔丁酯9a(1.0g,4.96mmol,上海毕得科技医药有限公司)溶解于二氯甲烷(20ml)中,加入戴斯
‑
马丁氧化剂(2.52g,5.94mmol),室温搅拌1小时。反应液减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物9b(800mg),产率80%。
[0425]
ms m/z(esi):200.2[m+1]
+
[0426]
第二步
[0427]
(r,z)
‑3‑
(二甲氨基)亚甲基)
‑2‑
甲基
‑4‑
氧代吡咯烷
‑1‑
羧酸叔丁酯9c
[0428]
将化合物9b(800mg,4.01mmol)溶解于n,n
‑
二甲基甲酰胺(20ml)中,加热升温至70℃反应1小时。反应液减压浓缩得到标题化合物9c(1g),产物不经纯化,直接用于下一步反应。
[0429]
ms m/z(esi):255.5[m+1]
+
[0430]
第三步
[0431]
(r)
‑4‑
甲基
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
羧酸叔丁酯9d
[0432]
将化合物9c(1g,3.40mmol)溶解于乙醇(20ml)中,加入水合肼(1.15g,19.52mmol,纯度85%),升温至70℃反应1小时。反应液减压浓缩,用柱层析以洗脱剂体系c纯化得到标题化合物9d(550mg),产率62%。
[0433]
ms m/z(esi):224.1[m+1]
+
[0434]
第四步
[0435]
(r)
‑2‑
(5,5
‑
二甲基
‑6‑
氧代
‑7‑
(2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
6,7
‑
二氢
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑4‑
甲基
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
羧酸叔丁酯9e
[0436]
将化合物9d(200mg,0.89mmol)溶解于n,n
‑
二甲基甲酰胺(5ml)中,加入化合物1b(293mg,0.89mmol)和碳酸铯(875mg,2.68mmol),升温至120℃反应6小时。反应液倒入水(30ml)中,用乙酸乙酯(30ml
×
2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物9e(60mg),产率13%。
[0437]
ms m/z(esi):515.2[m+1]
+
[0438]
第五步
[0439]
(r)
‑
5,5
‑
二甲基
‑4‑
(4
‑
甲基
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑7‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
5,7
‑
二氢
‑
6h
‑
吡咯并[2,3
‑
d]嘧啶
‑6‑
酮9f
[0440]
将化合物9e(60mg,0.12mmol)溶解于4m盐酸二氧六环(3ml)室温反应过夜,减压浓缩得到标题化合物9f(50mg),产物不经纯化直接用于下一步反应。ms m/z(esi):415.1[m+1]
+
[0441]
第六步
[0442]
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(r)
‑2‑
(5,5
‑
二甲基
‑6‑
氧代
‑7‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
6,7
‑
二氢
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑4‑
甲基
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)
‑3‑
氧代丙基)(异丙基)氨基甲酸叔丁酯9g
[0443]
将化合物9f(50mg,0.12mmol)溶解于n,n
‑
二甲基甲酰胺(3ml)中,加入化合物1f(41mg,0.12mmol)和2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(33mg,0.14mmol),二异丙基乙基胺(46mg,0.35mmol),室温反应20小时。反应液倒入水(30ml)中,用乙酸乙酯(30ml
×
2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩得到标题化合物9g(80mg),产物不经纯化直接用于下一步反应。ms m/z(esi):738.4[m+1]
+
[0444]
第七步
[0445]4‑
((r)
‑5‑
((s)
‑2‑
(4
‑
氯苯基)
‑3‑
(异丙基氨基)丙酰基)
‑4‑
甲基
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5,7
‑
二氢
‑
6h
‑
吡咯并[2,3
‑
d]嘧啶
‑6‑
酮9
[0446]
将化合物9g(80mg,0.11mmol)溶解于二氯甲烷(4ml)中,冰水浴下加入三氟乙酸(1ml),室温搅拌2小时。反应液减压浓缩,加入甲醇,水,碳酸钾调节ph值至碱性,搅拌过夜,反应液加水淬灭,二氯甲烷萃取(30ml
×
2),有机相无水硫酸钠干燥,减压浓缩后通过反相制备纯化(仪器型号:gilson 281色谱柱:sharpsil
‑
t,prep30
×
150mm;5μm;c18流动相:a
‑
水(0.1%三氟乙酸)b
‑
乙腈流速:30ml/min柱温:室温)得到目标化合物9(16mg),产率22%。
[0447]
ms m/z(esi):508.1[m+1]
+
[0448]1h nmr(500mhz,cd3od):δ8.56
‑
8.60(m,2h),7.44
‑
7.52(m,4h),5.28
‑
5.29(m,1h),4.29
‑
4.32(m,2h),3.68
‑
3.72(m,1h),3.48
‑
3.49(m,1h),3.28
‑
3.29(m,2h),1.63
‑
1.67(m,3h),1.61(s,3h),1.54(s,3h),1.30
‑
1.34(m,6h)。
[0449]
实施例10
‑
p1、10
‑
p2
[0450]
(s)
‑4‑
(5
‑
(3
‑
氨基
‑2‑
(4
‑
氯苯基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5,7
‑
二氢
‑
6h
‑
吡咯并[2,3
‑
d]嘧啶
‑6‑
酮10
‑
p1
[0451]
(r)
‑4‑
(5
‑
(3
‑
氨基
‑2‑
(4
‑
氯苯基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5,7
‑
二氢
‑
6h
‑
吡咯并[2,3
‑
d]嘧啶
‑6‑
酮10
‑
p2
[0452][0453]
第一步
[0454]
(2
‑
(4
‑
氯苯基)
‑3‑
(2
‑
(5,5
‑
二甲基
‑6‑
氧代
‑7‑
((2
‑
(三甲基甲硅烷基)乙氧基)甲基)
‑
6,7
‑
二氢
‑
5h
‑
吡咯并[2,3
‑
d]嘧啶
‑4‑
基)
‑
2,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
5(4h)
‑
基)
‑3‑
氧丙基)氨基甲酸叔丁酯10a
[0455]
将化合物1e(146mg,0.33mmol)溶解于n,n
‑
二甲基甲酰胺(5ml)中,加入化合物5a(100mg,0.33mmol)和2
‑
(7
‑
偶氮苯并三氮唑)
‑
四甲基脲六氟磷酸酯(127mg,0.33mmol),二异丙基乙基胺(130mg,1.0mmol),室温反应20小时。反应液倒入30ml水中,用乙酸乙酯(30ml
×
2)萃取,有机相用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩。用柱层析以洗脱剂体系c纯化得到标题化合物10a(180mg),产率79%。
[0456]
ms m/z(esi):682.1[m+1]
+
[0457]
第二步
[0458]
(s)
‑4‑
(5
‑
(3
‑
氨基
‑2‑
(4
‑
氯苯基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5,7
‑
二氢
‑
6h
‑
吡咯并[2,3
‑
d]嘧啶
‑6‑
酮10
‑
p1
[0459]
(r)
‑4‑
(5
‑
(3
‑
氨基
‑2‑
(4
‑
氯苯基)丙酰基)
‑
5,6
‑
二氢吡咯并[3,4
‑
c]吡唑
‑
2(4h)
‑
基)
‑
5,5
‑
二甲基
‑
5,7
‑
二氢
‑
6h
‑
吡咯并[2,3
‑
d]嘧啶
‑6‑
酮10
‑
p2
[0460]
将化合物10a(180mg,0.26mmol)溶解于二氯甲烷(3ml)中,加入三氟乙酸(3ml),室温搅拌2小时。反应液减压浓缩,加入甲醇(4ml)、水(1ml)和碳酸钾(36mg,2.63mmol),搅拌过夜,反应液加水淬灭,二氯甲烷(30ml
×
3)萃取,有机相用无水硫酸钠干燥,减压浓缩后通过反相制备纯化(仪器型号:gilson 281色谱柱:sharpsil
‑
t,prep30
×
150mm;5μm;c18流动相:a
‑
水(0.1%三氟乙酸)b
‑
乙腈流速:30ml/min柱温:室温)得到混合物(80mg)。
[0461]
通过手性制备纯化(仪器型号:gilson 281色谱柱:chiralpakig,5.0cm i.d.
×
25cm;10μm;c18流动相:hexane/ipa/hac=95/5/0.1(v/v/v)流速:60ml/min柱温:38℃)得到目标化合物(50mg,30mg),总收率68%。
[0462]
单一构型化合物(保留时间:8.38min):
[0463]
ms m/z(esi):452.1[m+1]
+
[0464]1h nmr(500mhz,cd3od):δ8.54
‑
8.59(m,2h),7.41
‑
7.46(m,4h),4.90(dd,1h),4.79(d,1h),4.65(dd,1h),4.37
‑
4.31(m,1h),4.24(t,1h),3.80
‑
3.70(m,1h),3.54
‑
3.43(m,1h),2.52(dd,1h),1.61(s,3h),1.60(dd,3h)。
[0465]
单一构型化合物(保留时间:11.60min):
[0466]
ms m/z(esi):452.1[m+1]
+
[0467]1h nmr(500mhz,cd3od):δ8.54
‑
8.59(m,2h),7.41
‑
7.46(m,4h),4.90(dd,1h),4.79(d,1h),4.65(dd,1h),4.37
‑
4.31(m,1h),4.24(t,1h),3.80
‑
3.70(m,1h),3.54
‑
3.43(m,1h),2.52(dd,1h),1.61(s,3h),1.60(dd,3h)。
[0468]
测试例:
[0469]
生物学评价
[0470]
测试例1、本公开化合物对akt1/akt2/akt3酶学实验评价
[0471]
以下方法用来测定本公开化合物在体外对akt1/akt2/akt3激酶活性的抑制作用。实验方法简述如下:
[0472]
akt1(invitrogen,p2999)、akt2(invitrogen,pv3184)和akt3(invitrogen,pv3185)的酶活性使用kinease
‑
stk s3 kit(cisbio,62st3pec)测定。首先用dmso将待测化合物从500μm开始进行3倍梯度稀释,共11个浓度点。将kit中的5
×
缓冲液稀释成1
×
缓冲液,并加入dtt(sigma,43816
‑
10ml)和mgcl2,使缓冲液中含1mm dtt和5mm mgcl2。用1
×
缓冲液将化合物稀释20倍待用。用1
×
缓冲液稀释akt1/akt2/akt3激酶得到酶溶液。用1
×
缓冲液稀释atp(invitrogen,pv3227)和kit中的s3
‑
biotin得到底物atp混合物溶液待用。在384孔板(corning,4513)中每孔加入2μl酶溶液和4μl化合物溶液,室温孵育30分钟,再加入4μl atp和s3
‑
biotin混合物溶液,室温孵育90分钟。akt1酶反应的条件为,酶终浓度为2nm,atp终浓度为10μm,s3
‑
biotin终浓度为2μm。akt2酶反应的条件为,酶终浓度为5nm,atp终浓度为10μm,s3
‑
biotin终浓度为2μm。akt3酶反应的条件为,酶终浓度为0.4nm,atp终浓度为45μm,s3
‑
biotin终浓度为2μm。使用kit中的detection buffer稀释s3
‑
cryptate和streptavidin
‑
xl665配制成detection溶液。孵育后,每孔加入10μl detection溶液,s3
‑
cryptate终浓度为stock稀释200倍,streptavidin
‑
xl665的终浓度为125nm。室温孵育60分钟,使用多功能微孔板检测仪(bmg labtech,pherastar fs)的htrf模块读取337nm激发,650nm和620nm发射的信号值,读数的比值乘以10000得到ratio值,用graphpad prism软件根据化合物的浓度和ratio值绘制量效曲线,并计算化合物抑制活性的ic
50
值。
[0473]
实验数据
[0474]
本公开化合物对akt1/akt2/akt3酶的抑制活性可通过以上的试验进行测定,测得的ic
50
值见表1。
[0475]
表1本公开化合物对akt1/akt2/akt3酶抑制的ic
50
值。
[0476][0477]
结论:本公开化合物对akt1/akt2/akt3酶均具有很好的抑制作用。
[0478]
药代动力学评价
[0479]
测试例2、本公开化合物的药代动力学测试
[0480]
1、摘要
[0481]
以大鼠为受试动物,应用lc/ms/ms法测定了大鼠灌胃给予实施例1化合物后不同时刻血浆中的药物浓度。研究本公开化合物在大鼠体内的药代动力学行为,评价其药动学特征。
[0482]
2、试验方案
[0483]
2.1试验药品
[0484]
实施例1化合物。
[0485]
2.2试验动物
[0486]
健康成年sd大鼠4只,雌雄各半,购自维通利华实验动物有限公司。
[0487]
2.3药物配制
[0488]
称取一定量药物,加入5%dmso、5%吐温80和90%生理盐水配置成无色澄明溶液。
[0489]
2.4给药
[0490]
sd大鼠禁食过夜后灌胃给药,给药剂量均为2mg/kg,给药体积均为10.0ml/kg。
[0491]
3.操作
[0492]
大鼠灌胃给药实施例1化合物,于给药前及给药后0.25,0.5,1.0,2.0,4.0,6.0,8.0,11.0,24.0小时由眼眶采血0.2ml,置于edta
‑
k2抗凝试管中,4℃、10000rpm离心1分钟,1h内分离血浆,于
‑
20℃保存,采血至离心过程在冰浴条件下操作,给药后2小时进食。
[0493]
测定不同浓度的药物灌胃给药后大鼠血浆中的待测化合物含量:取给药后各时刻的大鼠血浆20μl,加入甲醇400μl(其中含有100ng/ml内标:甲苯磺丁脲tolbutamide)沉淀
血浆样品,涡旋混合1分钟,离心7分钟(18000转/分钟)。将200μl上清液转移到96孔板中,3μl上清液进样用于lc/ms/ms分析。
[0494]
4、药代动力学参数结果
[0495]
本公开化合物的药代动力学参数如下:
[0496][0497]
结论:本公开化合物的药代吸收良好,具有明显的药代动力学优势。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1