一种泮托拉唑杂质的制备方法与流程
本发明涉及化合物合成技术,特别是涉及一种泮托拉唑杂质的制备方法。
技术背景
泮托拉唑(pantoprazole),常用其钠盐,临床上是一种质子泵抑制剂药物,抑制胃酸分泌。用于治疗活动性消化性溃疡反流性食管炎和卓一艾氏综合症。化学名为5-二氟甲氧基-2-[(3,4-二甲氧基-2-吡啶基)甲基]亚硫酰基-1h-苯并咪唑,分子量是:383.37,结构式为:
泮托拉唑钠(pantoprazolesodium)是由瑞士nycomedpharma公司研制开发,于1994年首次在德国和墨西哥上市,至今已在美国、英国、德国等20多个国家和地区上市。泮托拉唑钠是继奥美拉唑,兰索拉唑之后上市的第三代质子泵抑制剂(ppi),同其他质子泵抑制剂相比,在弱酸条件下稳定,在强酸条件下很快被激活,与其他药物相互作用小。泮托拉唑钠已成为胃溃疡系统疾病治疗的主流药物。
随着时代的进步以及科技水平的提高,人们对药品上市前必须对药品进行质量、安全性和效能科学评价的重要性等有了更加充分的认识,其中与药品质量密切相关的是药物所含杂质的控制。杂质往往与药品安全性有直接关系。各国药典都对药物杂质的含量和种类做了严格的规定。
本申请制备的泮托拉唑杂质(化合物vii),它是泮托拉唑合成过程中产生的一种最常见的杂质,化学名称为:5-(difluoromethoxy)-1-((3,4-dimethoxypyridin-2-yl)methyl)-2-(((3,4-dimethoxypyridin-2-yl)methyl)sulfinyl)-1h-benzo[d]imidazole,分子量为534.53,分子式为c25h26cl2n4o,结构式如下:
泮托拉唑杂质(化合物vii)比原料药泮托拉唑多一个二甲氧基吡啶结构,很有可能具比泮托拉唑更强的疗效。
journalofpharmaceuticalandbiomedicalanalysis45(2007)201–210在2004年报道了泮托拉唑杂质(化合物vii)的合成,文献中的部分合成路线如下图:
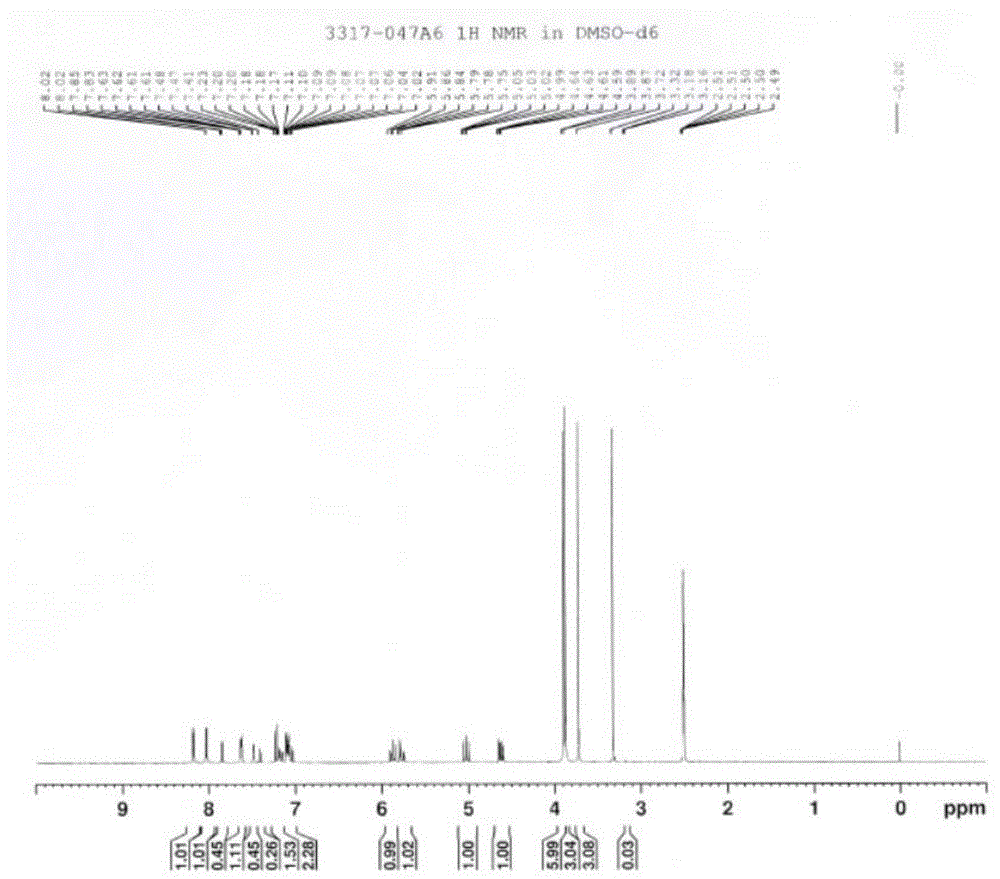
申请人参考上述文献,结果合成得到了混合物,journaloftheamericanchemicalsociety(2004),126(25),7800-7811在2004年报道了api甲基化的方法,文献提到其甲基化得到的是2个异构体的混合物(acsomega(2019),4(1),737-744报道的文献也可以证明),申请人也按文献尝试,还是得到了难以分离的异构体,混合物的核磁如附图1所示,8.20(d,1h),8.02(d,1h),7.85-7.50(m,2h),7.20-7.02(m,4h),5.91-5.75(m,2h),5.05-4.99(m,1h),4.64-4.59(m,1h),3.89-3.87(d,6h),3.72(s,3h),3.32(s,3h)。
技术实现要素:
发明目的:针对上述现有技术中关于泮托拉唑杂质的合成物质不单一的问题,本申请提供了一种高纯度、单一构型的泮托拉唑杂质的制备方法。
技术方案:本申请所述的一种泮托拉唑杂质的制备方法,合成路线为:
包括如下步骤:
(1)取化合物i溶于n,n-二甲基甲酰胺中,加入氢化钠和化合物ii2-氯甲基-3,4-二甲氧基吡啶盐酸盐,反应得化合物iii;
(2)取化合物iii溶于溶剂中,加入催化剂和还原剂,反应得化合物iv;
(3)取化合物iv悬浮于乙醇中,加入二硫化碳,碱,反应得化合物v;
(4)取化合物v和化合物ii悬浮于溶剂中,加入碱,反应得化合物vi;
(5)取化合物vi溶于有机溶剂中,加入氧化剂,反应提纯得到化合物vii;
步骤(1)中,所述化合物i为1当量,氢化钠和化合物ii均为1~2当量。所述反应温度为0~80℃,优选室温;反应时间为3~24小时,优选过夜。优选室温搅拌过夜反应。
步骤(2)中,所述的反应溶剂为甲苯、四氢呋喃、甲醇或乙醇,优选甲醇;所述催化剂选自二氧化铂、氢氧化钯、氯化钯、钯碳等,优选钯碳;所述还原剂选自铁粉、甲酸铵、锌粉、水合肼、氢气,优选水合肼。所述化合物iii为1当量,催化剂为0.05~0.5当量,还原剂为1~3当量。
步骤(2)中,所述的反应温度为20~100℃,优选75℃;所述的反应时间为0.5~4小时,优选1小时。
步骤(3)中,所述的碱选自碳酸钾、碳酸钠、氢氧化钾、氢氧化钠等,浓度在2%~8%,优选氢氧化钾。
步骤(3)中,所述化合物iv为1当量,碱用量为0.5~2当量。二硫化碳加入量为乙醇体积的2~35倍。
步骤(3)中,所述的反应时间为0.5~5小时,优选2小时;所述的反应温度为20~100℃,优选65℃。
步骤(4)中,所述的溶剂选自甲醇、乙醇、叔丁醇、正丁醇等,优选乙醇。
步骤(4)中,所用的碱选自氢化钠、碳酸钾、氢氧化钠、氢氧化钾等,优选氢氧化钠。所述碱浓度为浓度为1%~50%。进一步的,优选氢氧化钠水溶液,所述的氢氧化钠浓度为1%~50%,优选4%。
步骤(4)中,所述化合物v为1当量,化合物ii为和0.9~2当量,碱为1~3当量。所述的反应温度为0~80℃,优选室温;所述的反应时间为0.5~6小时,优选3小时。
步骤(5)中,所述的有机溶剂选自乙酸乙酯、甲醇、二氯甲烷、四氢呋喃等,优选二氯甲烷。
步骤(5)中,所述的氧化剂选自双氧水、次氯酸钠、间氯过氧苯甲酸、樟脑磺哑嗪等,优选间氯过氧苯甲酸。
步骤(5)中,所述化合物vi为1当量,所述氧化剂为0.8~1.5当量。所述的反应温度为-30~40℃,优选-10℃。
有益效果:本申请合成工艺设计合理,可操作性强;合成方法中使用的试剂简单易得,反应可以得到单一构型的化合物泮托拉唑杂质,通过hnmr、ms表征,其纯度可达99%以上,收率可达70%以上。本申请得到的泮托拉唑杂质可为原料药泮托拉唑的质量控制提供供试和对照样品,在药物申报具有重要的应用价值。
附图说明
图1为根据现有技术合成的混合物的核磁谱图;
图2为实施例1化合物iii的质谱谱图;
图3为实施例1化合物iii的核磁谱图;
图4为实施例2化合物iv的质谱谱图;
图5为实施例1化合物v的质谱谱图;
图6为实施例1化合物v的核磁谱图;
图7为实施例2化合物vii的质谱谱图;
图8为实施例3化合物vii的核磁谱图;
图9为实施例3化合物vii的液相谱图。
具体实施方式
下面结合具体实施例对本申请作出详细说明。
实施例1
如图1所示:泮托拉唑杂质的制备方法,具体包括以下步骤:
化合物iii的制备:取化合物i(4.00g,0.020mol)溶于40ml干燥dmf中,冰浴下加入氢化钠(1.60g,0.040mol),室温搅拌30min后,加入化合物ii(4.39g,0.020mol),室温搅拌过夜。加水调碱,析出固体抽滤得到化合物iii黄色固体6.75g,产率96.98%。质谱如图2所示,ms+:378。核磁如图3所示,9.14(br,1h),8.29(s,1h),8.00(s,1h),7.30(d,1h),7.09-7.07(s,1h),6.86-6.85(d,1h),6.60-6.26(m,1h),4.61-4.60(d,2h),3.94-3.92(m,6h)。
化合物iv的制备:取化合物iii(6.5g,0.018mol)溶于65.0ml甲醇中,加入水合肼(6.91g,0.136mol),钯碳(0.65g),75度搅拌1小时。冷却,抽滤除去钯碳,滤液旋干得到化合物iv类白色固体5.85g,产率98.32%。
化合物v的制备:取化合物iv(5.50g,0.017mol)悬浮于55.0ml乙醇中,加入氢氧化钾(0.48g,0.0085mol)和二硫化碳(16.5ml),65度搅拌2小时。旋干乙醇,加70ml水,用1n盐酸调节ph至5析出固体抽滤得到化合物v灰色固体5.95g,产率95.81%。质谱如图5所示,ms+:368,390。核磁如图6所示,12.88(br,1h),7.98-7.97(d,1h),7.37-6.95(m,5h),5.57(s,2h),3.89-3.88(d,6h)。
化合物vi的制备:取化合物v(5.80g,0.016mol)和化合物ii(3.54g,0.016mol)悬浮于116ml的乙醇中,加入4%氢氧化钠水溶液(31.7ml),室温反应6小时。加入200ml水,搅拌析出固体抽滤干燥得化合物vi白色固体6.58g,产率80.37%。
化合物vii的制备:取化合物vi(6.00g,0.012mol)溶于二氯甲烷(180.00ml)中,加入间氯过氧苯甲酸(2.53g,0.011mol),-10度反应1小时。加入10%硫代硫酸钠水溶液淬灭,用饱和碳酸钠水溶液(100ml)洗涤,无水硫酸钠干燥,用乙酸乙酯重结晶得到化合物vii白色固体5.15g,测hplc纯度为99.31%,产率83.33%。
实施例2
如图1所示:泮托拉唑杂质的制备方法,具体包括以下步骤:
化合物iii的制备:取化合物i(20.00g,0.098mol)溶于200ml干燥dmf中,冰浴下加入氢化钠(7.84g,0.196mol),室温搅拌30min后,加入化合物ii(21.96g,0.098mol),室温搅拌过夜。加水调碱,析出固体抽滤得到化合物iii黄色固体30.20g,产率86.75%。
化合物iv的制备:取化合物iii(30.00g,0.084mol)溶于300.0ml甲醇中,加入水合肼(31.88g,0.630mol),钯碳(3.00g),75度搅拌1小时。冷却,抽滤除去钯碳,滤液旋干得到化合物iv类白色固体26.87g,产率97.85%。质谱如图4所示,ms+:326。
化合物v的制备:取化合物iv(26.00g,0.080mol)悬浮于260.0ml乙醇中,加入氢氧化钾(2.28g,0.040mol)和二硫化碳(78.0ml),65度搅拌2小时。旋干乙醇,加300ml水,用1n盐酸调节ph至5析出固体抽滤得到化合物v灰色固体27.80g,产率94.69%。
化合物vi的制备:取化合物v(27.00g,0.073mol)和化合物iv(16.46g,0.073mol)悬浮于540ml的乙醇中,加入4%氢氧化钠水溶液(147.4ml),室温反应6小时。加入1000ml水,搅拌析出固体抽滤干燥得化合物vi白色固体26.80g,产率70.32%。
化合物vii的制备:取化合物vi(26.00g,0.050mol)溶于二氯甲烷(780.00ml)中,加入间氯过氧苯甲酸(10.35g,0.045mol),-10度反应1小时。加入10%硫代硫酸钠水溶液淬灭,用饱和碳酸钠水溶液(500ml)洗涤,无水硫酸钠干燥,用乙酸乙酯重结晶得到化合物vii白色固体20.12g,测hplc纯度为99.60%,产率75.07%。质谱如图7所示,ms+:535。
实施例3
如图1所示:泮托拉唑杂质的制备方法,具体包括以下步骤:
化合物iii的制备:取化合物i(137.00g,0.671mol)溶于1370ml干燥dmf中,冰浴下加入氢化钠(53.68g,1.342mol),室温搅拌30min后,加入化合物ii(150.36g,0.671mol),室温搅拌过夜。加水调碱,析出固体抽滤得到化合物iii黄色固体189.35g,产率79.40%。
化合物iv的制备:取化合物iii(189.00g,0.532mol)溶于1890.0ml甲醇中,加入水合肼(199.70g,3.990mol),钯碳(18.90g),75度搅拌1小时。冷却,抽滤除去钯碳,滤液旋干得到化合物iv类白色固体163.45g,产率94.45%。
化合物v的制备:取化合物iv(163.00g,0.501mol)悬浮于3260.0ml乙醇中,加入氢氧化钾(14.31g,0.250mol)和二硫化碳(4898.0ml),65度搅拌2小时。旋干乙醇,加3000ml水,用1n盐酸调节ph至5析出固体抽滤得到化合物v灰色固体160.23g,产率87.05%。
化合物vi的制备:取化合物v(160.00g,0.435mol)和化合物ii(97.47g,0.435mol)悬浮于3200ml的乙醇中,加入4%氢氧化钠水溶液(873.5ml),室温反应6小时。加入5000ml水,搅拌析出固体抽滤干燥得化合物vi白色固体156.34g,产率69.22%。
化合物vii的制备:取化合物vi(156.00g,0.301mol)溶于二氯甲烷(4680.00ml)中,加入间氯过氧苯甲酸(62.33g,0.271mol),-10度反应1小时。加入10%硫代硫酸钠水溶液淬灭,用饱和碳酸钠水溶液(4000ml)洗涤,无水硫酸钠干燥,用乙酸乙酯重结晶得到化合物vii白色固体118.26g,测hplc纯度为99.60%,产率73.54%。核磁如图8所示,8.24-8.22(d,1h),8.14-8.12(d,1h),7.59-7.58(d,1h),7.42-7.40(d,1h),7.10(d,1h),6.81-6.76(m,2h),6.68-6.30(m,1h),5.89-5.85(d,1h),5.77(d,1h),4.91-4.88(d,1h),3.91-3.88(m,12h)。液相如图9所示,rt:6.306min,99.60%。
- 还没有人留言评论。精彩留言会获得点赞!