一种带有导向基团的脂肪链烯烃的合成方法
1.本发明涉及有机化学技术领域,具体为一种带有导向基团的脂肪链烯烃的合成方法。
背景技术:
2.带有导向基团的惰性烯烃广泛应用于有机合成方法学的研究,全合成的研究,进而合成复杂的药物分子,通过导向基团的控制可以提高反应的区域选择性和化学选择性,这是一种强有力的工具来辅助惰性烯烃进行各种偶联反应,进而构建复杂的c
‑
c键、c
‑
n键、c
‑
s键、c
‑
o键等。
3.该化合物的化学名为2
‑
氰基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺,淡黄色固体。
4.目前已有的带有导向基团的惰性烯烃种类有限,并不能广泛应用,且通常所需反应条件较高,原料成本高,产率较低,导致作业过程中浪费较多,且后处理纯化较为困难。
5.因此,有必要提供一种带有导向基团的脂肪链烯烃的合成方法解决上述技术问题。
技术实现要素:
6.本发明的目的在于提供一种带有导向基团的脂肪链烯烃的合成方法,以解决上述背景技术中提出的问题。
7.为实现上述目的,本发明提供如下技术方案:一种带有导向基团的脂肪链烯烃的合成方法,包括以下步骤:
8.s1、在惰性气体氛围下,向含有磁子的干燥化学容器中加入存放于60%矿物油中的氢化钠和超干四氢呋喃,该溶液通过冰浴降到0℃,该温度下逐滴加入氰乙酸乙酯,0℃下反应20
‑
40分钟后加入3
‑
溴丙烯,该反应液加热到室温然后反应4
‑
8小时,待原料完全转化,向反应体系中加入15
‑
30毫升的水淬灭,再用15
‑
30毫升的乙酸乙酯萃取三次,将所有有机相收集,随后用30
‑
40毫升水洗两次,30
‑
40毫升饱和食盐水洗一次,有机相用无水硫酸镁干燥,通过过滤除去不溶性物质,通过旋蒸除去乙酸乙酯,之后通过柱层析得到3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯;
9.s2、向得到的3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯中加入1.0
‑
1.5当量的2m氢氧化钠的水溶液,按每摩尔3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯加2
‑
3毫升乙醇溶液,室温下反应15
‑
20小时,反应结束后用旋蒸除去乙醇,旋干之后的反应液中加入和乙醇相同体积的水,再用相同体积的二氯甲烷洗三遍,向水相中加2m的盐酸酸化,调ph至4.0,再用二氯甲烷萃取后得到3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸;
10.s3、得到的3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸加入0.8当量8
‑
氨基喹啉,1.0当量hatu,1.55当量吡啶,用超干二氯甲烷作溶剂,在室温下反应16
‑
24小时至原料完全转化,用旋蒸除去溶剂二氯甲烷,加入60
‑
100ml乙酸乙酯溶解反应液,加入80
‑
120ml饱和碳酸氢钠,萃取三次,混合有机相加入80
‑
120ml饱和食盐水洗涤,旋干后通过快速柱层析得到纯产物2
‑
氰
基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺。
11.进一步的,在所述步骤s1中,所述惰性气氛所用惰性气体为氩气。
12.进一步的,在所述步骤s1中,所述氰乙酸乙酯、3
‑
溴丙烯、氢化钠的摩尔比为3:1:1。
13.进一步的,在所述步骤s1中,所述氢化钠的质量分数为10%
‑
30%,氰乙酸乙酯的质量分数为40%
‑
80%,3
‑
溴丙烯的质量分数为10%
‑
30%,3
‑
溴丙烯与超干四氢呋喃的摩尔体积比为10mmol:40ml。
14.进一步的,在所述s2步骤中,所述3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯的质量分数为1%
‑
3%,所述1.0
‑
1.5当量的2m氢氧化钠的水溶液中的氢氧化钠为5%
‑
10%,水溶液为70%
‑
95%,3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯与四氢呋喃的摩尔体积比为10mmol:15ml。
15.进一步的,在所述步骤s3中,所述3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸的质量分数为15%
‑
30%、8
‑
氨基喹啉的质量分数为10%
‑
25%、hatu的质量分数为15%
‑
30%、吡啶的质量分数为20%
‑
50%,8
‑
氨基喹啉与超干二氯甲烷的摩尔体积比为1mmol:3ml。
16.进一步的,在所述步骤s2中,所述3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯、氢氧化钠、水的摩尔比为1:4:50。
17.进一步的,在所述步骤s2中,所加入的2m盐酸的体积为5ml。
18.进一步的,在所述步骤s3中,所用二氯甲烷为超干二氯甲烷,所述快速柱层析的流动相为摩尔比为1:3的石油醚:乙酸乙酯硅胶色谱柱纯化。
19.与现有技术相比,本发明的有益效果是:该带有导向基团的脂肪链烯烃的合成方法,通过该方法得到了一种新的化合物,该化合物可以广泛用于各类惰性烯烃的偶联反应,该合成路线具有很好的官能团兼容性,且反应条件温和,原料便宜易得,产率高,后处理纯化简单的特点。
附图说明
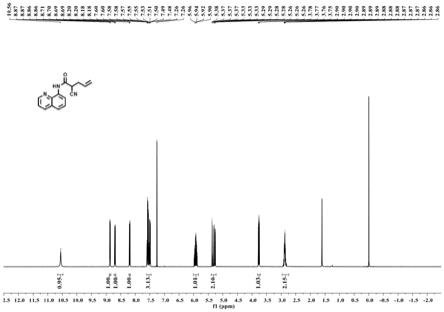
20.图1为本发明提供的带有导向基团的脂肪链烯烃的合成方法中的2
‑
氰基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺的氢谱图。
具体实施方式
21.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
22.实施例一:
23.s1、在惰性气体氛围下,向含有磁子的干燥化学容器中加入存放于60%矿物油中的氢化钠和超干四氢呋喃,该溶液通过冰浴降到0℃,该温度下逐滴加入氰乙酸乙酯,0℃下反应20
‑
40分钟后加入3
‑
溴丙烯,该反应液加热到室温然后反应4
‑
8小时,待原料完全转化,向反应体系中加入15
‑
30毫升的水淬灭,再用15
‑
30毫升的乙酸乙酯萃取三次,将所有有机相收集,随后用30
‑
40毫升水洗两次,30
‑
40毫升饱和食盐水洗一次,有机相用无水硫酸镁干燥,通过过滤除去不溶性物质,通过旋蒸除去乙酸乙酯,之后通过柱层析得到3
‑
氰基
‑2‑
氧
杂己基
‑5‑
烯酸乙酯;
24.s2、向得到的3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯中加入1.0
‑
1.5当量的2m氢氧化钠的水溶液,按每摩尔3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯加2
‑
3毫升乙醇溶液,室温下反应15
‑
20小时,反应结束后用旋蒸除去乙醇,旋干之后的反应液中加入和乙醇相同体积的水,再用相同体积的二氯甲烷洗三遍,向水相中加2m的盐酸酸化,调ph至4.0,再用二氯甲烷萃取后得到3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸;
25.s3、得到的3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸加入0.8当量8
‑
氨基喹啉,1.0当量hatu,1.55当量吡啶,用超干二氯甲烷作溶剂,在室温下反应16
‑
24小时至原料完全转化,用旋蒸除去溶剂二氯甲烷,加入60
‑
100ml乙酸乙酯溶解反应液,加入80
‑
120ml饱和碳酸氢钠,萃取三次,混合有机相加入80
‑
120ml饱和食盐水洗涤,旋干后通过快速柱层析得到纯产物2
‑
氰基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺。
26.在所述步骤s1中,所述惰性气氛所用惰性气体为氩气。
27.在所述步骤s1中,所述氰乙酸乙酯、3
‑
溴丙烯、氢化钠的摩尔比为3:1:1。
28.在所述步骤s1中,所述氢化钠的质量分数为10%
‑
30%,氰乙酸乙酯的质量分数为40%
‑
80%,3
‑
溴丙烯的质量分数为10%
‑
30%,3
‑
溴丙烯与超干四氢呋喃的摩尔体积比为10mmol:40ml。
29.在所述s2步骤中,所述3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯的质量分数为1%
‑
3%,所述1.0
‑
1.5当量的2m氢氧化钠的水溶液中的氢氧化钠为5%
‑
10%,水溶液为70%
‑
95%,3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯与四氢呋喃的摩尔体积比为10mmol:15ml。
30.在所述步骤s3中,所述3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸的质量分数为15%
‑
30%、8
‑
氨基喹啉的质量分数为10%
‑
25%、hatu的质量分数为15%
‑
30%、吡啶的质量分数为20%
‑
50%,8
‑
氨基喹啉与超干二氯甲烷的摩尔体积比为1mmol:3ml。
31.在所述步骤s2中,所述3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯、氢氧化钠、水的摩尔比为1:4:50。
32.在所述步骤s2中,所加入的2m盐酸的体积为5ml。
33.在所述步骤s3中,所用二氯甲烷为超干二氯甲烷,所述快速柱层析的流动相为摩尔比为1:3的石油醚:乙酸乙酯硅胶色谱柱纯化。
34.hatu是一种效果较好的缩合剂,3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸和8
‑
氨基喹啉为主要缩合物,吡啶承担一部分碱的作用,超干二氯甲烷为溶解性较好的溶剂,8
‑
氨基喹啉、3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸、hatu和吡啶这四种物质以1:1.3:1.3:2的比例搭配,可以达到很好的反应效果,其中3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸是可以选择4
‑
戊烯酸、2
‑
甲基
‑4‑
戊酸、2
‑
苯基
‑4‑
戊酸等烯酸及其衍生物进行替换的;
[0035]2‑
氰基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺的沸点是599.1
±
45.0℃,密度是1.244
±
0.06g/cm3,酸度系数是10.54
±
0.43;
[0036]2‑
甲基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺的沸点是450.3
±
28.0℃,密度是1.136
±
0.06g/cm3,酸度系数是12.92
±
0.43。
[0037]
将2
‑
氰基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺与芳基卤化物反应,加入2.5mol%
‑
10mol%催化剂、5mol%
‑
20mol%配体、3
‑
5当量醋酸锂以及20
‑
25当量的水,加入醇类溶剂如2,3
‑
丁二醇,在100℃
‑
140℃下反应,最终实现偶联反应。
[0038]
实施例二:
[0039]
s1、在惰性气体氛围下,向含有磁子的干燥化学容器中加入存放于60%矿物油中的氢化钠和超干四氢呋喃,该溶液通过冰浴降到0℃,该温度下逐滴加入氰乙酸乙酯,0℃下反应20
‑
40分钟后加入3
‑
溴丙烯,该反应液加热到室温然后反应4
‑
8小时,待原料完全转化,向反应体系中加入15
‑
30毫升的水淬灭,再用15
‑
30毫升的乙酸乙酯萃取三次,将所有有机相收集,随后用30
‑
40毫升水洗两次,30
‑
40毫升饱和食盐水洗一次,有机相用无水硫酸镁干燥,通过过滤除去不溶性物质,通过旋蒸除去乙酸乙酯,之后通过柱层析得到3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯;
[0040]
s2、向得到的3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯中加入1.0
‑
1.5当量的2m氢氧化钠的水溶液,按每摩尔3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯加2
‑
3毫升乙醇溶液,室温下反应15
‑
20小时,反应结束后用旋蒸除去乙醇,旋干之后的反应液中加入和乙醇相同体积的水,再用相同体积的二氯甲烷洗三遍,向水相中加2m的盐酸酸化,调ph至4.0,再用二氯甲烷萃取后得到3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸;
[0041]
s3、得到的3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸加入0.8当量8
‑
氨基喹啉,1.0当量hatu,1.55当量吡啶,用超干二氯甲烷作溶剂,在室温下反应16
‑
24小时至原料完全转化,用旋蒸除去溶剂二氯甲烷,加入60
‑
100ml乙酸乙酯溶解反应液,加入80
‑
120ml饱和碳酸氢钠,萃取三次,混合有机相加入80
‑
120ml饱和食盐水洗涤,旋干后通过快速柱层析得到纯产物2
‑
氰基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺。
[0042]
在所述步骤s1中,所述惰性气氛所用惰性气体为氩气。
[0043]
在所述步骤s1中,所述氰乙酸乙酯、3
‑
溴丙烯、氢化钠的摩尔比为3:1:1。
[0044]
在所述步骤s1中,所述氢化钠的质量分数为10%
‑
30%,氰乙酸乙酯的质量分数为40%
‑
80%,3
‑
溴丙烯的质量分数为10%
‑
30%,3
‑
溴丙烯与超干四氢呋喃的摩尔体积比为10mmol:40ml。
[0045]
在所述s2步骤中,所述3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯的质量分数为1%
‑
3%,所述1.0
‑
1.5当量的2m氢氧化钠的水溶液中的氢氧化钠为5%
‑
10%,水溶液为70%
‑
95%,3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯与四氢呋喃的摩尔体积比为10mmol:15ml。
[0046]
在所述步骤s3中,所述3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸的质量分数为15%
‑
30%、8
‑
氨基喹啉的质量分数为10%
‑
25%、hatu的质量分数为15%
‑
30%、吡啶的质量分数为20%
‑
50%,8
‑
氨基喹啉与超干二氯甲烷的摩尔体积比为1mmol:3ml。
[0047]
在所述步骤s2中,所述3
‑
氰基
‑2‑
氧杂己基
‑5‑
烯酸乙酯、氢氧化钠、水的摩尔比为1:4:50。
[0048]
在所述步骤s2中,所加入的2m盐酸的体积为5ml。
[0049]
在所述步骤s3中,所用二氯甲烷为超干二氯甲烷,所述快速柱层析的流动相为摩尔比为1:3的石油醚:乙酸乙酯硅胶色谱柱纯化。
[0050]
hatu是一种效果较好的缩合剂,3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸和8
‑
氨基喹啉为主要缩合物,吡啶承担一部分碱的作用,超干二氯甲烷为溶解性较好的溶剂,8
‑
氨基喹啉、3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸、hatu和吡啶这四种物质以1:1.3:1.3:2的比例搭配,可以达到很好的反应效果,其中3
‑
氰基
‑2‑
氧杂己烷
‑5‑
烯酸是可以选择4
‑
戊烯酸、2
‑
甲基
‑4‑
戊酸、2
‑
苯基
‑4‑
戊酸等烯酸及其衍生物进行替换的;
[0051]2‑
氰基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺的沸点是599.1
±
45.0℃,密度是1.244
±
0.06g/cm3,酸度系数是10.54
±
0.43;
[0052]2‑
甲基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺的沸点是450.3
±
28.0℃,密度是1.136
±
0.06g/cm3,酸度系数是12.92
±
0.43。
[0053]
将2
‑
氰基
‑
n
‑
(喹啉
‑8‑
基)戊
‑4‑
烯酰胺与烷基卤化物反应,加入2.5mol%
‑
10mol%光催化剂、5mol%
‑
20mol%配体、3.0当量的碳酸铯,加入能将其溶解的溶剂如以40:1比例的2
‑
(异丙基氨)乙醇和甲醇混合溶液,在光照下反应,实现在自由基偶联反应。
[0054]
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1