一种富马酸伏诺拉生中间体的制备方法与流程
本发明属于医药技术领域,具体涉及一种富马酸伏诺拉生中间体的制备方法。
背景技术:
富马酸伏诺拉生(vonoprazanfumarate)是由日本武田公司开发的一种新型胃酸分泌抑制剂,曾用代号tak-438。
2015-02-01,武田薬品工業株式会社在日本上市了富马酸伏诺拉生片,规格10mg和20mg,商品名为
2019-12-18,takedapharmaceuticalcompanylimited公司的富马酸伏诺拉生片首次进口中国,商品名
vonoprazanfumarate的化学名称为1-[5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-基]-n-甲基甲胺富马酸盐,cas号:1260141-27-2、881681-00-1(伏诺拉生),结构式如下所示:
专利cn10130022a中介绍了原研合成路线,具体合成路线如下:从起始物料到成品需要5步,在反应过程中中需要过柱纯化,总收率不足5%,而且在整个反应过程中需要使用超低温-78℃等苛刻条件对设备要求高。
武田公司在专利cn102421753a中对原研合成路线进行改进,以5-(2-氟苯基)-2-氯-1h-吡咯-3-腈为原料,经过四步反应得到产品,总收率约为39%。
其中,化合物i为制备富马酸伏诺拉生的关键中间体,由化合物ii制备化合物i的过程中,容易发生过度还原反应,生成化合物ix,并进一步和化合物i反应生成化合物x和化合物xi。在后处理过程中,这些副产物会以油状物的方式析出,包裹大量化合物i从而导致收率降低,而且会残留至正常析出的化合物i中导致纯度降低。相关的副产物的结构式如下所示:
专利cn102421753a中制备化合物i的后处理步骤简单包括如下步骤:(1)过滤反应液,除去不溶性的雷尼镍等物质,用乙酸乙酯淋洗滤饼;(2)向得到的滤液中滴加氢氧化钠溶液调节其ph至7~8,分液,有机相采用碳酸氢钠溶液和氯化钠溶液洗涤;(3)洗涤后得到的有机相用盐酸调节ph其至3.0~3.5,再次分液,得到的有机相采用氯化钠溶液洗涤,然后减压浓缩;(4)得到的浓缩物升温至65~70℃,再降温至45~55℃,继续搅拌1h,再降温至15~25℃,滴加正庚烷,滴加完毕后,降温至0~10℃,搅拌析晶1h,过滤干燥,得到中间体。上述化合物i的精制过程收率达到80%左右,但是仍然存在如下缺点:在步骤(2)中,将反应液的ph调节至7~8,分液时,对副产物化合物x和化合物xi的分离效果差,得到的有机相中化合物i的纯度较低;在步骤(3)中,浓缩前将ph调节至3.0~3.5时,浓缩过程中反应液中化合物i会降解,收率和纯度显著降低;在步骤(4)中,在添加正庚烷析晶过程中,副产物会以油状物的方式析出,包裹大量化合物i从而导致收率偏低。
因此,针对中间体(化合物i)制备过程中后处理困难,收率和纯度低的问题,提供一种高纯度和高收率的富马酸伏诺拉生中间体(化合物i)的制备方法显得尤为重要。
技术实现要素:
本发明的目的是在现有技术的基础上,提供一种富马酸伏诺拉生中间体的制备方法,针对反应过程中副产物多和产物易降解的难题,在后处理的过程中,严格控制浓缩前后反应液的ph,可以有效去除副产物,降低浓缩时目标产物的降解,以及避免在正庚烷析晶的过程中,出现黑色油状物质,从而导致目标产物的收率和纯度偏低。采用本发明的制备方法显著提高了整个反应过程中目标产物的收率和纯度,降低成本,适合大规模生产。
本发明的技术方案如下:
一种富马酸伏诺拉生中间体的制备方法,它包括以下步骤:
(1)向氢化釜中加入化合物ii、四氢呋喃、乙酸、水和雷尼镍,搅拌均匀后,在氢气氛围下,于温度为20~60℃和压力为0~0.6mpa的条件下进行化学反应,待反应完成后,过滤,乙酸乙酯洗涤淋洗滤饼,得到反应液;具体合成路线如下:化合物ii化学名称为5-(2-氟苯基)-1h-吡咯-3-腈,化合物i化学名称为5-(2-氟苯基)-1h-吡咯-3-甲醛。
(2)将步骤(1)中得到的反应液与乙酸乙酯混合,在20~30℃的条件下滴加氢氧化钠水溶液,调节其ph至4.5~6.5,分液弃去水相,有机相采用氯化钠水溶液洗涤,再次分液弃去水相。
(3)将步骤(2)中洗涤后得到的有机相在30~60℃的条件下浓缩至干。
(4)将步骤(3)中得到的浓缩物与乙酸乙酯混合,滴加氢氧化钠水溶液,调节其ph至7.5~9.5,分液弃去水相。
(5)向步骤(4)中得到的有机相中滴加正庚烷,滴加完毕后再降温至0~30℃进行搅拌析晶,过滤,干燥,即得中间体化合物i。
对于本发明而言,由化合物ii制备化合物i时,一般在氢化釜中进行,采用氮气置换氢化釜中的空气,以四氢呋喃、醋酸和水为溶剂,氢源为氢气,雷尼镍为催化剂。
在步骤(1)中,化合物ii与四氢呋喃的质量体积比为1:3~10g/ml,可以但不局限于1:3g/ml、1:4g/ml、1:5g/ml、1:6g/ml、1:7g/ml、1:8g/ml、1:9g/ml或1:10g/ml,在一种优选方案中,化合物ii与四氢呋喃的质量体积比为1:4g/ml。
在步骤(1)中,化合物ii与乙酸的质量体积比为1:4~10g/ml,可以但不局限于1:4g/ml、1:5g/ml、1:6g/ml、1:7g/ml、1:8g/ml、1:9g/ml或1:10g/ml,在一种优选方案中,化合物ii与乙酸的质量体积比为1:6g/ml。
在步骤(1)中,化合物ii与水的质量体积比为1:1~10g/ml,可以但不局限于1:1g/ml、1:2g/ml、1:3g/ml、1:4g/ml、1:5g/ml、1:6g/ml、1:7g/ml、1:8g/ml、1:9g/ml或1:10g/ml,在一种优选方案中,化合物ii与水的质量体积比为1:2g/ml。
在步骤(1)中,化合物ii与雷尼镍的质量比为1:0.4~0.8,可以但不局限于1:0.4、1:0.5、1:0.6、1:0.7或1:0.8,在一种优选方案中,化合物ii与雷尼镍的质量比为1:0.6。
在步骤(1)中,待反应完成后,过滤,乙酸乙酯淋洗滤饼,淋洗滤饼时,在化合物ii与乙酸乙酯的质量体积比1:1~3g/ml,可以但不局限于1:1g/ml、1:1.2g/ml、1:1.5g/ml、1:1.7g/ml、1:1.8g/ml、1:1.9g/ml、1:2.0g/ml、1:2.2g/ml、1:2.5g/ml、1:3.0g/ml,在一种优选方案中,化合物ii与乙酸乙酯的质量体积比1:1.8g/ml。
在步骤(1)中,由化合物ii制备化合物i的过程中,反应温度为20~60℃,可以但不局限于20℃、30℃、40℃、50℃或60℃,在一种优选方案中,反应温度为20~30℃。
以氢气作为氢源,在反应过程中,控制压力为0~0.6mpa,优选地,控制压力为0~0.1mpa。
进一步地,反应时间为4~10小时,可以但不局限于4小时、5小时、6小时、7小时、8小时或10小时。
在步骤(1)中,在反应的过程中,搅拌速度为100~400r/min,优选地,搅拌速度为200~300r/min。
乙酸乙酯的投加量需要严格控制,过高或者过低影响萃取时的效率,在步骤(2)中,步骤(1)中化合物ii与乙酸乙酯的质量体积比1:10~30g/ml,可以但不局限于1:10g/ml、1:12g/ml、1:15g/ml、1:18g/ml、1:20g/ml、1:25g/ml或1:30g/ml,在一种优选方案中,步骤(1)中化合物ii与乙酸乙酯的质量体积比1:15~20g/ml。
在步骤(4)中,步骤(1)中化合物ii与乙酸乙酯的质量体积比1:4~15g/ml,可以但不局限于1:4g/ml、1:5g/ml、1:6g/ml、1:7g/ml、1:8g/ml、1:10g/ml、1:12g/ml或1:15g/ml,在一种优选方案中,步骤(1)中化合物ii与乙酸乙酯的质量体积比1:6~10g/ml。
在析晶过程中正庚烷的投加量需要严格控制,过高或者过低影响析晶效率,在步骤(5)中,步骤(1)中化合物ii与正庚烷的质量体积比1:5~25g/ml,可以但不局限于1:5g/ml、1:8g/ml、1:10g/ml、1:12g/ml、1:15g/ml、1:20g/ml或1:25g/ml,在一种优选方案中,步骤(1)中化合物ii与正庚烷的质量体积比1:10~20g/ml。
对于本发明而言,由化合物ii制备化合物i的过程中,即使严格控制反应过程中的物料投加量和反应温度,也容易发生过度还原反应生成化合物ix,并进一步和化合物i反应生成化合物x和化合物xi。在后处理过程中,副产物(化合物ix、化合物x和化合物xi)会以油状物的方式析出,包裹大量化合物i从而导致收率降低,而且会残留至正常析出的化合物i中导致纯度降低,所以在制备化合物i时,一般收率和纯度低,成本高,影响后续反应过程中伏诺拉生产品的收率和品质。
本发明在步骤(1)中制备含化合物i的反应液,然后针对得到的反应液进行后处理,特别是在步骤(2)中,将步骤(1)中得到的反应液与乙酸乙酯混合,滴加氢氧化钠水溶液,调节其ph至4.5~6.5,分液弃去水相,氯化钠水溶液洗涤,洗涤后得到的有机相进行浓缩。由于化合物i的结构不稳定,在浓缩的过程中,化合物i容易降解,以及化合物x和化合物xi难以去除,从而导致收率和纯度偏低。而本申请在浓缩前,严格控制反应液的ph至4.5~6.5,降低化合物i在浓缩的过程中发生降解的概率,提高了产物的收率和纯度。在一种优选方案中,在步骤(2)中,滴加氢氧化钠水溶液,调节其ph至5.0~6.0,在浓缩的过程中,化合物i几乎不降解,从而提高了收率和纯度。
若浓缩前,反应液的ph偏低,例如,滴加氢氧化钠水溶液,调节其ph至3.0~4.0,在浓缩的过程中,化合物i特别容易降解,使得收率和纯度显著降低。若浓缩前,反应液的ph偏高,例如,滴加氢氧化钠水溶液,调节其ph至7.0~8.0,在浓缩的过程中,化合物x和化合物xi难以去除,使得副产物的含量高,同时化合物i会部分降解,导致后处理过程中收率和纯度偏低。
在一种优选方案中,在步骤(2)中,滴加氢氧化钠水溶液,调节其ph时,氢氧化钠水溶液的浓度为5~20wt%,可以但不局限于5wt%、8wt%、10wt%、15wt%、20wt%,进一步地,氢氧化钠水溶液的浓度为10~20wt%。
在一种优选方案中,在步骤(3)中,步骤(2)中洗涤后得到的有机相在40~60℃的条件下浓缩至干。例如,浓缩至干时的温度为40~50℃或50~60℃。
对于本发明而言,在步骤(3)中,有机相浓缩后,在加入正庚烷析晶之前,也需要严格控制反应液的ph。浓缩后,添加氢氧化钠水溶液调节其ph,当ph偏低时,例如,在步骤(4)中,浓缩物与乙酸乙酯混合,滴加氢氧化钠水溶液,调节其ph至5.0~6.0,分液弃去水相,然后将得到的有机相滴加正庚烷进行搅拌析晶时,副产物会以油状物的方式析出,包裹大量化合物i从而导致收率偏低。当ph偏高时,例如,滴加氢氧化钠水溶液,调节其ph至10.0~11.0,在滴加正庚烷进行搅拌析晶时,由于碱性太强,导致化合物i部分降解,从而导致收率和纯度降低。
本发明在步骤(4)中,添加氢氧化钠水溶液,调节其ph至7.5~9.5,可以避免在滴加正庚烷进行搅拌析晶时:因ph过低时,副产物会以油状物的方式析出,包裹大量化合物i;以及ph过高时,化合物i发生部分降解,从而导致化合物i的收率和纯度偏低的问题。在一种优选方案中,在步骤(4)中,添加氢氧化钠水溶液,调节其ph至8.0~9.0。
在步骤(4)中,滴加氢氧化钠水溶液,调节其ph时,氢氧化钠水溶液的浓度为0.4~15wt%,可以但不局限于0.4wt%、0.8wt%、1wt%、3wt%、5wt%、7wt%、10wt%、12wt%或15wt%,进一步地,氢氧化钠水溶液的浓度为1~10wt%。
在步骤(5)中,搅拌析晶时温度为0~30℃,优选地,搅拌析晶时温度为0~10℃。
进一步地,搅拌析晶时时间为0.5~2小时,优选为1小时。
采用本发明的技术方案,优势如下:
本发明提供的富马酸伏诺拉生中间体的制备方法,针对反应过程中副产物多和产物易降解的难题,在后处理的过程中,严格控制浓缩前后反应液的ph,在浓缩前控制其ph至4.5~6.5,在浓缩的过程中,目标产物的稳定性好不易降解,也可以有效去除副产物化合物x和化合物xi,提高了收率和纯度;在浓缩后控制其ph至7.5~9.5,在滴加正庚烷析晶的过程中,不会出现黑色油状物质包裹目标产物,从而降低收率和纯度。采用本发明的制备方法,产物的收率和纯度高,收率达到90%,纯度达到99%,后处理简单,成本低,适合大规模生产。
附图说明
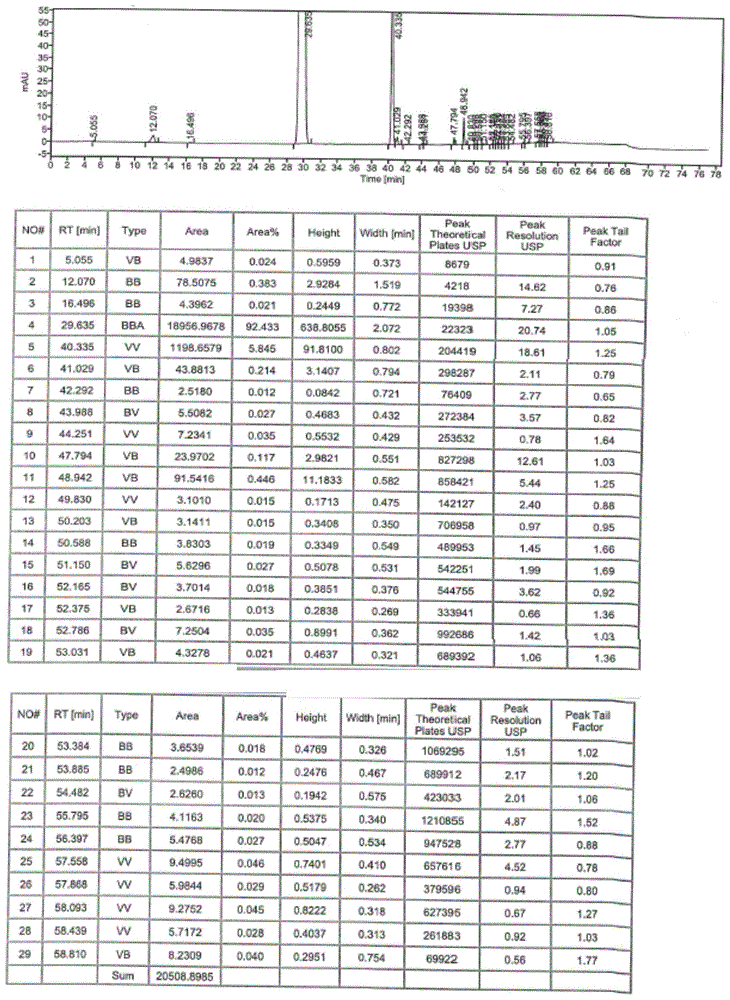
图1是实施例1中制备的氢化反应液的hplc图;
图2是实施例2中制备的中间体化合物i的hplc图;
图3是实施例3中制备的中间体化合物i的hplc图;
图4是实施例4中制备的中间体化合物i的hplc图;
图5是对比例1中制备的中间体化合物i的hplc图;
图6是对比例2中制备的中间体化合物i的hplc图;
图7是对比例3中制备的中间体化合物i的hplc图;
图8是对比例4中制备的中间体化合物i的hplc图;
图9是对比例5中制备的中间体化合物i的hplc图;
图10是对比例6中制备的中间体化合物i的hplc图;
图11是对比例7中制备的中间体化合物i的hplc图。
具体实施方式
通过以下实施例并结合附图对本发明的制备方法作进一步的说明,但这些实施例不对本发明构成任何限制。
实施例1:化合物i反应液制备
向2l氢化釜中加入化合物ii(56.00g,0.30mol)、四氢呋喃224.0ml、乙酸336.0ml、纯化水112.0ml和雷尼镍33.60g,氮气置换三次后加氢气,开启搅拌,控制搅拌速度为200~300r/min,控制氢气压力为0~0.1mpa,控温20~30℃的条件下反应5h,tlc监控化合物ii物料点消失(pe:ea=2:1),过滤,滤饼用乙酸乙酯100.0ml淋洗,收集滤液,均分8份,冷藏备用。hplc纯度92.43%。
实施例2:化合物i的精制(方案c)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯105.0ml,控温20~30℃的条件下滴加10%氢氧化钠水溶液,调节其ph至5.0,分液,弃去水相,有机相用5%氯化钠水溶液洗涤两次,每次105.0ml,分液,再次弃去水相。将得到的有机相转移至旋转蒸发仪中,在40~50℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入42.0ml乙酸乙酯,搅拌溶解,滴加1%氢氧化钠水溶液,调节其ph至8.0。分液,弃去水相。有机相转移至250ml三口瓶中,在20~30℃的条件下,滴加正庚烷77.0ml,有大量固体析出,再降温至0~10℃,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)6.44g,收率为90.54%,纯度为99.66%。
实施例3:化合物i的精制(方案c)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯140.0ml,控温20~30℃的条件下滴加20%氢氧化钠水溶液,调节其ph至6.0,分液,弃去水相,有机相用5%氯化钠水溶液洗涤两次,每次105.0ml,分液,再次弃去水相。将得到的有机相转移至旋转蒸发仪中,在50~60℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入70.0ml乙酸乙酯,搅拌溶解,滴加10%氢氧化钠水溶液,调节其ph至9.0。分液,弃去水相。有机相转移至250ml三口瓶中,在20~30℃的条件下,滴加正庚烷140.0ml,有大量固体析出,再降温至20~30℃,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)6.42g,收率为90.26%,纯度为99.53%。
实施例4:化合物i的精制(方案c)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯140.0ml,控温20~30℃的条件下滴加20%氢氧化钠水溶液,调节其ph至6.5,分液,弃去水相,有机相用5%氯化钠水溶液洗涤两次,每次105.0ml,分液,再次弃去水相。将得到的有机相转移至旋转蒸发仪中,在50~60℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入70.0ml乙酸乙酯,搅拌溶解,滴加10%氢氧化钠水溶液,调节其ph至9.0。分液,弃去水相。有机相转移至250ml三口瓶中,在20~30℃的条件下,滴加正庚烷140.0ml,有大量固体析出,再降温至20~30℃,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)6.40g,收率为89.98%,纯度为99.31%。
对比例1:化合物i的精制(方案a)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯105.0ml,控温20~30℃的条件下滴加10%氢氧化钠水溶液,调节其ph至5.0,分液,弃去水相,有机相用5%氯化钠水溶液洗涤两次,每次105.0ml,分液,再次弃去水相。将得到的有机相转移至旋转蒸发仪中,在40~50℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入42.0ml乙酸乙酯,搅拌溶解,滴加1%氢氧化钠水溶液,调节其ph至5.0。分液,弃去水相。有机相转移至250ml三口瓶中,在20~30℃的条件下,滴加正庚烷77.0ml,有大量固体析出,同时瓶壁上有黑色油状物,再降温至0~10℃,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)4.05g,收率为56.94%,纯度为97.58%。
对比例2:化合物i的精制(方案b)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯105.0ml,控温20~30℃的条件下滴加10%氢氧化钠水溶液,调节其ph至8.0,分液,弃去水相,有机相用5%氯化钠水溶液洗涤两次,每次105.0ml,分液,再次弃去水相。将得到的有机相转移至旋转蒸发仪中,在40~50℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入42.0ml乙酸乙酯,搅拌溶解,滴加1%氢氧化钠水溶液,调节其ph至8.0。分液,弃去水相。有机相转移至250ml三口瓶中,在20~30℃的条件下,滴加正庚烷77.0ml,有大量固体析出,再降温至0~10℃,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)5.12g,收率为71.98%,纯度为93.86%。
对比例3:化合物i的精制(方案d)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯105.0ml,控温20~30℃的条件下滴加10%氢氧化钠水溶液,调节其ph至4.0,分液,弃去水相,有机相用5%氯化钠水溶液洗涤两次,每次105.0ml,分液,再次弃去水相。将得到的有机相转移至旋转蒸发仪中,在40~50℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入42.0ml乙酸乙酯,搅拌溶解,滴加1%氢氧化钠水溶液,调节其ph至8.0。分液,弃去水相。有机相转移至250ml三口瓶中,在20~30℃的条件下,滴加正庚烷77.0ml,有大量固体析出,再降温至0~10℃,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)5.33g,收率为74.94%,纯度为93.81%。
对比例4:化合物i的精制(方案e)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯105.0ml,控温20~30℃的条件下滴加10%氢氧化钠水溶液,调节其ph至5.0,分液,弃去水相,有机相用5%氯化钠水溶液洗涤两次,每次105.0ml,分液,再次弃去水相。将得到的有机相转移至旋转蒸发仪中,在40~50℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入42.0ml乙酸乙酯,搅拌溶解,滴加1%氢氧化钠水溶液,调节其ph至10.5。分液,弃去水相。有机相转移至250ml三口瓶中,在20~30℃的条件下,滴加正庚烷77.0ml,有大量固体析出,再降温至0~10℃,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)6.18g,收率为86.89%,纯度为93.08%。
对比例5:化合物i的精制(方案f,参照专利cn102421753a中实施例3)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯105.0ml,控温20~30℃的条件下滴加20%氢氧化钠水溶液,调节其ph至7.5,分液,弃去水相,有机相用5%碳酸氢钠水溶液105.0ml洗涤一次,再用5%氯化钠水溶液105.0ml洗涤一次,分液,再次弃去水相。将得到的有机相中加入纯化水105.0ml,在20~30℃的条件下滴加6mol/l盐酸水溶液调节其ph至3.0,搅拌过夜。分液后,将有机相用5%氯化钠水溶液105.0ml洗涤一次,分液,弃去水相。然后,将有机相转移至旋转蒸发仪中,在40~50℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入42.0ml乙酸乙酯,搅拌溶解,升温至60~70℃,再降温至45~55℃,搅拌1h,继续降温至15~25℃,滴加正庚烷77.0ml,有大量固体析出,同时瓶壁上有黑色油状物,然后降温至0~10℃,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)4.92g,收率为69.17%,纯度为92.90%。
对比例6:化合物i的精制(调整浓缩后添加乙酸乙酯的比例)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯105.0ml,控温20~30℃的条件下滴加10%氢氧化钠水溶液,调节其ph至5.0,分液,弃去水相,有机相用5%氯化钠水溶液洗涤两次,每次105.0ml,分液,再次弃去水相。将得到的有机相转移至旋转蒸发仪中,在40~50℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入140.0ml乙酸乙酯,搅拌溶解,滴加1%氢氧化钠水溶液,调节其ph至8.0。分液,弃去水相。有机相转移至250ml三口瓶中,在20~30℃的条件下,滴加正庚烷77.0ml,有大量固体析出,再降温至0~10℃,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)5.70g,收率为80.14%,纯度为99.57%。
对比例7:化合物i的精制(调整精制析晶温度)
取实施例1中的1份滤液,加入至500ml三口瓶中,向其中加入乙酸乙酯105.0ml,控温20~30℃的条件下滴加10%氢氧化钠水溶液,调节其ph至5.0,分液,弃去水相,有机相用5%氯化钠水溶液洗涤两次,每次105.0ml,分液,再次弃去水相。将得到的有机相转移至旋转蒸发仪中,在40~50℃的条件下浓缩至冷凝处无液体流下,再将得到的浓缩物转移至250ml三口瓶中,加入42.0ml乙酸乙酯,搅拌溶解,滴加1%氢氧化钠水溶液,调节其ph至8.0。分液,弃去水相。有机相转移至250ml三口瓶中,在20~30℃的条件下,滴加正庚烷77.0ml,有大量固体析出,在40~50℃条件下,搅拌析晶1h,过滤,所得固体在40~50℃下真空干燥6~10h,得黄色固体(中间体,化合物i)5.94g,收率为83.51%,纯度为99.65%。
针对上述实施例和对比例,摘录相关的实验数据如下表1所示:
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可能对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!
- Josiphos类手性二茂铁膦配体的合成方法
- (S)-1-(2-羟基-1-苯乙基)硫脲修饰的Mn-Anderson型杂多酸催化剂、制备方法及其应用
- (S)-1-(1-苯乙基)硫脲修饰的Cr-Anderson型杂多酸催化剂、制备方法及其应用
- (R)-1-(1-苯乙基)硫脲修饰的Cr-Anderson型杂多酸催化剂、制备方法及其应用
- 以机械增强来提高纤维素生物质的微生物转化的方法
- (S)-1-(2-羟基-1-苯乙基)硫脲修饰的Cr-Anderson型杂多酸催化剂、制备方法及其应用
- 一种微电解-Fenton内循环固定床反应器的制造方法
- 一种光学活性中间体n-叔丁氧羰基-2-氨基-8-壬烯酸二环己胺盐的合成方法
- 一种光学活性中间体n-叔丁氧羰基-2-氨基-8-壬烯酸二环己胺盐的合成方法
- 一种仿过氧化物酶材料及其应用