一种高效制备丹酚酸B和紫草酸的方法
一种高效制备丹酚酸b和紫草酸的方法
技术领域
1.本发明涉及一种高效制备丹酚酸b和紫草酸的方法。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.滇丹参(salvia yunnanensis radix)为唇形科植物云南鼠尾草(salvia yunnanensis c.h.wright)的干燥根,又称紫丹参,在云南、贵州地区有悠久的药用历史,始载于明代《滇南本草》中。滇丹参的活性作用与丹参(salvia miltiorrhizabge.)相似,其生物活性主要是对循环系统的影响,如改善微循环和血液循环功能、提升心脏冠脉血通量并修复心脏功能、降血脂、预防动脉粥样硬化,另外还有肝脏保护、治疗胃溃疡、镇痛、抗氧化、抗菌抗炎、抗肿瘤等。另有研究表明,滇丹参有显著的抑制血小板凝集的作用,且抑制效果明显优于丹参。滇丹参中的酚酸类化合物主要起到抗炎、抗氧化、抗血栓以及心血管保护的作用,以丹酚酸a~k为代表,其中丹酚酸b含量最高。然而,发明人研究发现,丹酚酸b与紫草酸的结构较为类似,采用普通色谱法难以同时获得高纯度的丹酚酸b与紫草酸。
技术实现要素:
4.为了解决现有技术的不足,本发明的目的是提供一种高效制备丹酚酸b和紫草酸的方法,采用该方法能够同时获得丹酚酸b与紫草酸,纯度均在95%以上。
5.ph区带逆流色谱是一种液
‑
液色谱技术,根据化合物的pka和极性的差异实现成分的分离,该技术具有分离效率高、重现性好、进样量大等优点。本发明采用首先以各种溶剂体系(例如石油醚
–
乙酸乙酯
–
甲醇
–
水、乙酸乙酯
–
甲醇
–
水、乙酸乙酯
–
正丁醇
–
水、乙酸乙酯
–
水、氯仿
–
甲醇
–
水)进行尝试,但是难以实现丹酚酸b和紫草酸的分离提取,当采用叔丁基甲醚
–
乙腈
–
水(4:1:5,v/v)作为溶剂系统,结果发现该溶剂系统可以实现丹酚酸b与紫草酸的分离提取,但是分离提取时间较长,仅洗脱丹酚酸b就需要9h,由于分离提取时间较长,造成ph区带逆流色谱分离提取丹酚酸b和紫草酸困难。
6.为了实现上述目的,本发明的技术方案为:
7.一方面,一种高效制备丹酚酸b和紫草酸的方法,采用ph区带逆流色谱对含有丹酚酸b和紫草酸的样品进行分离纯化;ph区带逆流色谱的溶剂系统由叔丁基甲醚、水组成,叔丁基甲醚和水的体积比为1:0.9~1.1,溶剂系统的上相溶液作为固定相,溶剂系统的下相溶液作为流动相,固定相中添加酸,流动相中添加碱。
8.本发明经过研究发现以叔丁基甲醚
–
乙腈
–
水(4:1:5,v/v/v)作为溶剂系统时,样品在乙腈中的溶解度比在水和叔丁基甲醚中的溶解度都要好,而乙腈对叔丁基甲醚的亲和度更好,导致样品保留时间较长。因而本发明重新调整溶剂系统,经过试验发现,采用该溶剂系统能够使得丹酚酸b和紫草酸在13h即可完成分离,大大降低了丹酚酸b和紫草酸的分
离时间。
9.同时,经过进一步研究发现,固定相的酸浓度和流动相的碱浓度也能够降低丹酚酸b和紫草酸的分离时间,当固定相中的酸浓度4.5~5.5mm、流动相中的碱浓度为59~61mm时,丹酚酸b和紫草酸的分离时间能够缩短至9h。
10.另一方面,一种上述方法在提取滇丹参中丹酚酸b和紫草酸的应用。
11.由于滇丹参中的丹酚酸b含量较高,且存在紫草酸,采用上述方法用于滇丹参的提取,能够有效将滇丹参中的丹酚酸b和紫草酸同时提取出来。
12.第三方面,一种滇丹参中丹酚酸b和紫草酸的提取方法,步骤如下:
13.(1)从滇丹参提取获得滇丹参总酚酸提取物;
14.(2)采用ph区带逆流色谱滇丹参总酚酸提取物进行分离纯化获得含有丹酚酸b和紫草酸的馏分;
15.(3)采用ph区带逆流色谱对将含有丹酚酸b和紫草酸的馏分进行分离纯化;
16.其中,步骤(3)中ph区带逆流色谱的溶剂系统由叔丁基甲醚、水组成,叔丁基甲醚和水的体积比为1:0.9~1.1,溶剂系统的上相溶液作为固定相,溶剂系统的下相溶液作为流动相,固定相中添加酸,流动相中添加碱。
17.本发明在对滇丹参中酚酸类化合物提取时,发现采用一次ph区带逆流色谱难以对滇丹参中的丹酚酸b和紫草酸分离,因而本发明在一次ph区带逆流色谱分离纯化的基础上再进行一次ph区带逆流色谱分离纯化,而经过多种溶剂体系的选择发现,仅有叔丁基甲醚
–
乙腈
–
水(4:1:5,v/v/v)溶剂体系能够实现对丹酚酸b和紫草酸的分离,但是分离时间极长,经过试验发现,采用叔丁基甲醚和水作为溶剂体系不仅能够完全分离丹酚酸b和紫草酸,而且分离时间大大缩短。
18.本发明的有益效果为:
19.1.本发明采用叔丁基甲醚和水作为ph区带逆流色谱的溶剂系统,不仅能够将丹酚酸b和紫草酸完全分离,而且能够使丹酚酸b和紫草酸的分离速率加快,降低丹酚酸b和紫草酸的分离时间。
20.2.本发明采用ph区带逆流色谱分离丹酚酸b和紫草酸,不仅能够滇丹参中提取出大量的丹酚酸b和紫草酸,而且获得的丹酚酸b和紫草酸的纯度较高,可达95%以上。
附图说明
21.构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
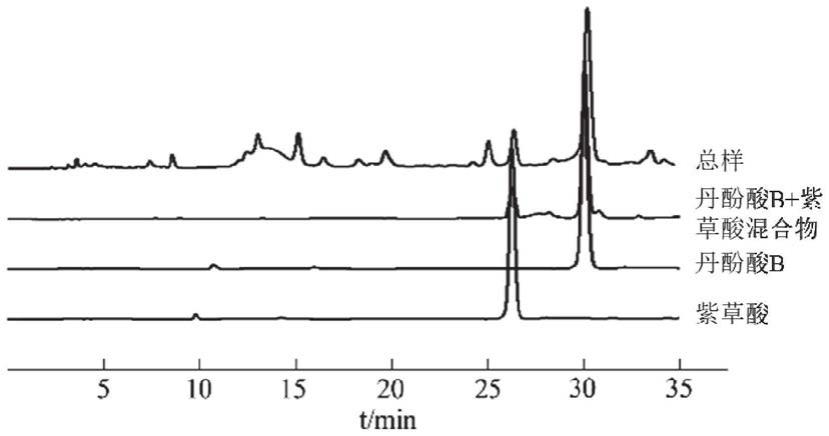
22.图1为本发明实施例中滇丹参总酚酸提取物与经ph
‑
zrccc分离得到的纯组分的高效液相色谱(hplc)分析图;
23.图2为本发明实施例中ph
‑
zrccc的分离富集图;
24.图3为本发明实施例中ph
‑
zrccc的分离图。
具体实施方式
25.应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常
理解的相同含义。
26.需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
27.丹酚酸b的化学结构为
28.紫草酸的化学结构为
29.鉴于ph区带逆流色谱的溶剂体系对丹酚酸b和紫草酸的分离效果及分离效率影响较大,本发明提出了一种高效制备丹酚酸b和紫草酸的方法。
30.本发明的一种典型实施方式,提供了一种高效制备丹酚酸b和紫草酸的方法,采用ph区带逆流色谱对含有丹酚酸b和紫草酸的样品进行分离纯化;ph区带逆流色谱的溶剂系统由叔丁基甲醚、水组成,叔丁基甲醚和水的体积比为1:0.9~1.1,溶剂系统的上相溶液作为固定相,溶剂系统的下相溶液作为流动相,固定相中添加酸,流动相中添加碱。
31.本发明经过研究发现以叔丁基甲醚
–
乙腈
–
水(4:1:5,v/v/v)作为溶剂系统时,样品在乙腈中的溶解度比在水和叔丁基甲醚中的溶解度都要好,而乙腈对叔丁基甲醚的亲和度更好,导致样品保留时间较长。因而本发明重新调整溶剂系统,经过试验发现,采用该溶剂系统能够使得丹酚酸b和紫草酸在13h即可完成分离,大大降低了丹酚酸b和紫草酸的分离时间。
32.将叔丁基甲醚和水混合均匀,静止分液,分液后的上相为固定相,分液后的下相作为流动相。
33.在一些实施例中,固定相中的酸浓度4.5~5.5mm,流动相中的碱浓度为59~61mm。经过实验发现,该条件下,分离丹酚酸b和紫草酸的效率进一步提高,从而进一步缩短丹酚酸b和紫草酸的分离时间。
34.在一些实施例中,所述酸为三氟乙酸,所述碱为氨水。
35.本发明的另一种实施方式,提供了一种上述方法在提取滇丹参中丹酚酸b和紫草酸的应用。
36.本发明的第三种实施方式,提供了一种滇丹参中丹酚酸b和紫草酸的提取方法,步骤如下:
37.(1)从滇丹参提取获得滇丹参总酚酸提取物;
38.(2)采用ph区带逆流色谱滇丹参总酚酸提取物进行分离纯化获得含有丹酚酸b和
紫草酸的馏分;
39.(3)采用ph区带逆流色谱对将含有丹酚酸b和紫草酸的馏分进行分离纯化;
40.其中,步骤(3)中ph区带逆流色谱的溶剂系统由叔丁基甲醚、水组成,叔丁基甲醚和水的体积比为1:0.9~1.1,溶剂系统的上相溶液作为固定相,溶剂系统的下相溶液作为流动相,固定相中添加酸,流动相中添加碱。
41.在一些实施例中,步骤(3)中,固定相中的酸浓度4.5~5.5mm,流动相中的碱浓度为59~61mm。经过实验发现,该条件下,分离丹酚酸b和紫草酸的效率进一步提高,从而进一步缩短丹酚酸b和紫草酸的分离时间。
42.在一些实施例中,步骤(3)中,所述酸为三氟乙酸,所述碱为氨水。
43.在一些实施例中,从滇丹参提取获得滇丹参总酚酸提取物的过程为:采用乙醇提取法对滇丹参进行提取,提取后的液体采用石油醚进行一次萃取,萃取后的水相调节ph至1.5~2.5,然后添加乙酸乙酯进行二次萃取,对二次萃取的乙酸乙酯相进行浓缩获得滇丹参总酚酸提取物。
44.本发明所述的一次萃取、二次萃取中的“一次”、“二次”仅表明不同的萃取过程,并不是对萃取次数的限定。
45.在一种或多种实施例中,将滇丹参加入乙醇水溶液,在65~75℃条件下进行回流提取,将提取液进行浓缩,再加水复溶,然后采用石油醚进行一次萃取。其中,乙醇水溶液的浓度55~65%体积分数。滇丹参与乙醇水溶液的料液比为1:7.5~8.5,kg:l。回流提取的次数为2~4次,每次回流提取的时间为1.5~2.5h。
46.在一种或多种实施例中,二次提取的次数为3~7次。
47.ph区带逆流色谱分离提取的过程为:将固定相输送至分离柱,将滇丹参总酚酸提取物的溶液注入分离柱,然后将流动相输送持续输送至分离柱进行分离提取。固定相输送至分离柱的流速为25~35ml/min。流动相的流速为1.5~2.5ml/min。收集馏分的时间为每次(瓶)收集6min。ph区带逆流色谱分离提取中,紫外检测器的检测波长为252~256nm。
48.在一些实施例中,步骤(2)中,ph区带逆流色谱的溶剂系统,包括石油醚、乙酸乙酯、乙腈和水,其中,石油醚、乙酸乙酯、乙腈和水的体积比为1.46~1.54:2.46~2.54:1:4.6~5.4。将石油醚、乙酸乙酯、乙腈和水混合均匀,静止分液,分液后的上相为固定相,分液后的下相作为流动相。当固定相中三氟乙酸的浓度为9~11mm、流动相中氨水的浓度为29~31mm时,分离提取的效果更为优异。
49.为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
50.以下实施例中采用的试剂若未进行特殊说明,均为市售获得。滇丹参药材购于云南省新平县。
51.实施例
52.样品的提取与制备:
53.称取滇丹参药材3kg并粉碎,使用60%乙醇在70℃下回流提取3次,每次2h,料液比为1:8(w/v)。将提取液过滤,50℃下减压浓缩至约800ml,并用水复溶至1000ml。提取液使用等量石油醚萃取三次,弃去上层,在水相加入盐酸将提取液ph调至2,然后使用等量乙酸乙酯萃取5次。合并乙酸乙酯相,减压浓缩,得滇丹参总酚酸提取物136g。
54.两相溶剂系统与样品溶液的制备:
55.根据两相溶剂系统,将各溶剂按比例分别量取并依次加入到分液漏斗中。振荡后静置12h,分出上、下两相。上相作为固定相加入一定浓度的三氟乙酸,下相作为流动相加入一定浓度的氨水。将上、下两相超声脱气2min,作为ph
‑
zrccc分离使用的溶剂系统。
56.将样品放入试管中,使用酸化后的上相溶液和等量未碱化的下相超声溶解,且总体积不超过20ml,制得ph
‑
zrccc分离使用的样品溶液。
57.ph区带逆流色谱的分离
58.将上相溶液以30ml/min的流速泵入,直至充满分离柱,然后将样品溶液通过进样环注入分离柱,将分离柱调为正转800rpm,同时将下相溶液以2ml/min的流速泵入。将紫外检测器的检测波长调为254nm,以6min/瓶的间隔收集馏分,并使用ph计检测各瓶馏分的ph值。分离过程结束后,使用空气压缩机将柱内剩余液体吹入到量筒中,计算固定相的保留率。最后使用水和乙醇依次对分离柱进行清洗。
59.hplc分析与结构鉴定
60.滇丹参总酚酸提取物与经ph
‑
zrccc分离得到的纯组分的hplc分析条件为compass c18色谱柱(250
×
4.6mm,5μm),流动相为乙腈(a)
–
0.1%甲酸水溶液(b):0
–
10min,14%a;10
–
20min,14%
–
17%a;20
–
35min,17%
–
30%a。流速1.0ml/min,检测波长280nm,进样量10μl。
61.ph
‑
zrccc分离得到的纯组分使用hplc
‑
esi
‑
qtof
‑
ms和
13
c
‑
nmr、1h
‑
nmr对纯组分进行结构鉴定。
62.hplc分析结果
63.图1所示为滇丹参总酚酸提取物与经ph
‑
zrccc分离得到的纯组分的hplc分析图,在280nm下使用峰面积归一化法进行计算,丹酚酸b和紫草酸在总酚酸粗提物中的峰面积比分别为32.12%、5.52%。
64.两相溶剂系统的优化
65.首先使用石油醚
–
乙酸乙酯
–
乙腈
–
水(1.5:2.5:1:5,v/v/v/v)作为分离溶剂系统,下相加入氨水(30mm),上相加入三氟乙酸(10mm),对2g滇丹参进行分离。结合hplc分析发现,如图2所示,由于丹酚酸b和紫草酸在该体系中色谱峰重叠较大,没有明显分离趋势,因此将两化合物的馏分合并,减压浓缩,冷冻干燥,得到样品1共1.30g,用于ph
‑
zrccc二次分离。
66.对于样品1的ph
‑
zrccc分离,首先使用了石油醚
–
乙酸乙酯
–
甲醇
–
水、乙酸乙酯
–
甲醇
–
水、乙酸乙酯
–
正丁醇
–
水、乙酸乙酯
–
水、氯仿
–
甲醇
–
水等体系进行了一系列尝试,均很难实现化合物iv和v的分离。根据ito提出的溶剂系统,再次尝试了叔丁基甲醚
–
乙腈
–
水(4:1:5,v/v/v)溶剂系统(上相加入三氟乙酸(10mm),下相加入氨水(10mm)),对样品1进行分离。结果发现该溶剂系统可以实现两化合物的分离,但洗脱时间较长,仅第一个化合物v的洗脱就持续了9h,因此如何缩短洗脱时间成为了问题关键。在实验发现,样品在乙腈中的溶解度比在水和叔丁基甲醚中的溶解度都要好,而在叔丁基甲醚
–
乙腈
–
水(4:1:5,v/v/v)体系中,乙腈对叔丁基甲醚的亲和度更好。所以,由于上述溶剂系统中乙腈的存在使样品大量的分布在固定相是导致样品保留时间较长的主要因素。因此,实验去掉了溶剂系统中的乙腈,使样品更多溶解于下相,然后使用叔丁基甲醚
–
水(1:1,v/v)体系,上相加入三氟乙酸
(10mm),下相加入氨水(10mm),对样品1进行分离,结果两化合物在13h完成分离。基于上述实验,本实施例对溶剂系统中的保留酸和洗脱碱的浓度进行了极限的调整,将保留酸浓度减少到5mm,洗脱碱浓度增加至60mm,并再次使用叔丁基甲醚
–
水(1:1,v/v)溶剂系统对1.30g的样品1进行了分离,ph
‑
zrccc分离图如图3所示。丹酚酸b和紫草酸在9h内实现了成功分离,两区带平台界限明显,纯度分别为95.6%和95.0%。经冷冻干燥后,得到丹酚酸b 259.9mg,紫草酸28.75mg。
67.化合物的结构鉴定
68.根据esi
‑
ms、1h
‑
nmr和
13
c
‑
nmr对化合物i
‑
iii进行结构鉴定,具体数据如下:
69.丹酚酸b:准分子离子峰为m/z 717[m
–
h]
–
,提示相对分子量为718,二级质谱碎片为m/z 519[m
–
salvianic acid a]
–
。1h
‑
nmr(dmso
‑
d6,400mhz)δ(ppm):7.14(1h,s,h
‑
5),6.82(1h,s,h
‑
6),6.19(1h,s,h
‑
8),6.23(1h,d,h
‑2’
),6.80
‑
6.51(8h,h
‑5’
,6’,2”,5”,6”,2
”’
,5
”’
,6
”’
),5.77(1h,s,7
”’
),4.22(1h,s,h
‑
8”),4.83(2h,s,h
‑8’
,8
”’
).
13
c
‑
nmr(dmso
‑
d6,100mhz)δ(ppm):125.6(c
‑
1),125.6(c
‑
2),146.1(c
‑
3),147.9(c
‑
4),119.8(c
‑
5),121.9(c
‑
6),144.0(c
‑
7),117.3(c
‑
8),166.5(c
‑
9),129.6(c
‑1’
),116.8(c
‑2’
),146.1(c
‑3’
),145.9(c
‑4’
),116.0(c
‑5’
),123.3(c
‑6’
),37.7(c
‑7’
),76.6(c
‑8’
),173.1(c
‑9’
),132.2(c
‑
1”),113.2(c
‑
2”),145.5(c
‑
3”),145.5(c
‑
4”),115.6(c
‑
5”),117.0(c
‑
6”),86.6(c
‑
7”),56.7(c
‑
8”),172.4(c
‑
9”),130.0(c
‑1”’
),116.3(c
‑2”’
),144.3(c
‑3”’
),144.1(c
‑4”’
),119.8(c
‑5”’
),123.3(c
‑6”’
),37.3(c
‑7”’
),77.5(c
‑8”’
),170.9(c
‑9”’
)。
[0070]
紫草酸:准分子离子峰为m/z 537[m
–
h]
–
,提示相对分子量为538,二级质谱碎片为m/z 493[m
–
h
–
co2]
–
和m/z 295[m
–
h
–
co2–
salvianic acid a]
–
。1h
‑
nmr(dmso
‑
d6,400mhz)δ(ppm):7.13(1h,d,j=8.0hz,h
‑
6),8.00(1h,d,j=16.0hz,h
‑
7),6.19(1h,d,j=16.0hz,h
‑
8),6.75
‑
6.66(5h,m,h
‑
5,2’,2”,5”,6”),6.51(1h,d,j=8.0hz,h
‑5’
),6.37(1h,d,j=8.0hz,h
‑6’
),2.88(1h,d,j=12.0hz,h
‑7’
a),2.72(1h,d,j=12.0hz,h
‑7’
b),4.58(1h,t,j=24.0,12.0hz,h
‑8’
),5.82(1h,d,j=4.0hz,h
‑
7”),4.05(1h,d,j=4.0hz,h
‑
8”).
13
c
‑
nmr(dmso
‑
d6,100mhz)δ(ppm):123.1(c
‑
1),129.7(c
‑
2),147.2(c
‑
3),143.7(c
‑
4),118.6(c
‑
5),119.5(c
‑
6),142.0(c
‑
7),116.4(c
‑
8),166.3(c
‑
9),131.0(c
‑1’
),117.4(c
‑2’
),145.8(c
‑3’
),144.2(c
‑4’
),116.4(c
‑5’
),119.5(c
‑6’
),37.8(c
‑7’
),76.7(c
‑8’
),173.1(c
‑9’
),133.5(c
‑
1”),113.6(c
‑
2”),146.3(c
‑
3”),145.7(c
‑
4”),118.5(c
‑
5”),115.9(c
‑
6”),88.7(c
‑
7”),60.1(c
‑
8”),174.5(c
‑
9”)。
[0071]
本发明使用溶剂系统石油醚
–
乙酸乙酯
–
乙腈
–
水(1.5:2.5:1:5,v/v/v/v)(上相加入三氟乙酸(10mm),下相加入氨水(30mm))从滇丹参总酚酸粗提物中获得丹酚酸b和紫草酸的馏分,然后使用溶剂系统叔丁基甲醚
–
水(1:1,v/v)获得259.9mg丹酚酸b和28.75mg紫草酸,纯度均高于95%。
[0072]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1