烃可溶性类盐氢化物催化剂及其制备方法与流程
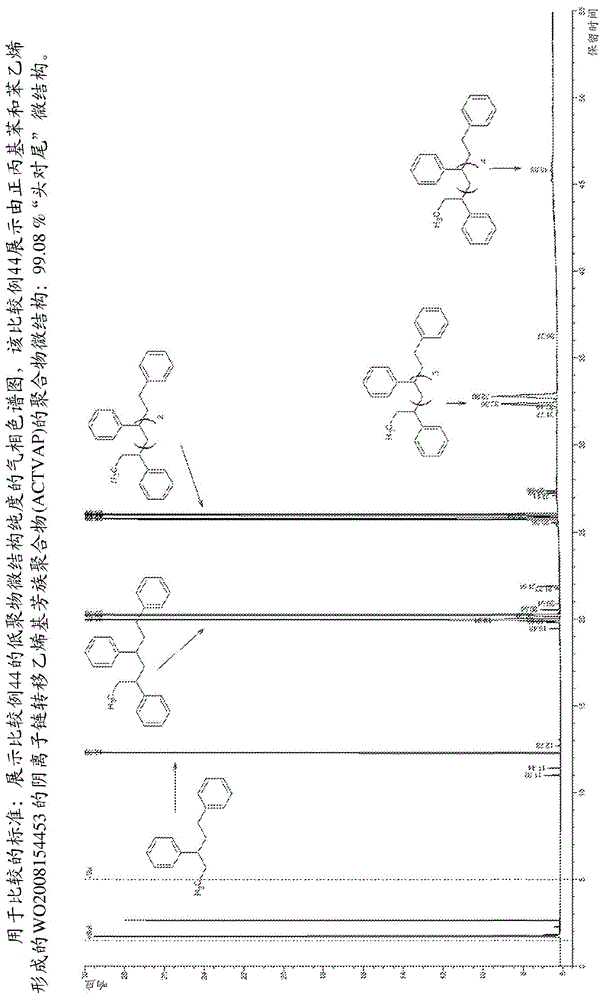
本申请为分案申请,其母案的申请日为2017年4月4日、申请号为201780034718.4、发明名称为“用于氢介导的类盐氢化物引发的阴离子链转移聚合的方法和烃可溶性类盐氢化物催化剂以及由其生成的聚合物分布组合物”。本发明涉及通过新型聚合条件形成氢介导的类盐氢化物(salinehydride)引发的阴离子聚合物分布的方法,其中分子氢是链转移剂,并且锂氨基醇盐络合的类盐氢化物(loxsh)通过使类盐氢化物加成到阴离子可聚合的烃单体中形成阴离子聚合物链引发物质;所有这些都以非常高效至高度高效的催化循环发生,其中动力学链长分布由氢介导或以其他方式通过氢气与单体的相对进料速率设定。本发明还涉及聚苯乙烯组合物,其具有大大改善的微结构,而没有共产物聚合物链分布。本发明还涉及可用于进行本发明的氢介导的类盐氢化物引发的聚合的新型烃可溶性类盐氢化物催化剂和试剂组合物。本发明还涉及由二甲基氨基乙醇(又名二甲基乙醇胺)、烷基锂试剂和分子氢形成的烃可溶性氢化锂催化剂和试剂组合物,还涉及催化剂形成方法,该催化剂在氢介导的苯乙烯阴离子聚合(hmaps)中的用途以及低不对称性和高“头对尾”微结构的所得低分子量聚苯乙烯分布。
背景技术:
:低分子量-mw<<4000道尔顿-聚苯乙烯组合物可用于诸如聚合物滑爽试剂(参见epo741147)的最终用途或用作进一步合成衍生化的底物前体的各种应用中。这种合成衍生化需要芳族亲电取代反应(参见:us8217120b2“官能化苯乙烯低聚物和聚合物(functionalizedstyreneoligomersandpolymers)”)。聚苯乙烯的阴离子链转移聚合提供了经济效益,这归因于在形成低分子量聚苯乙烯组合物时由于在形成链转移引发剂和催化剂中使用的有机锂试剂和其他碱土金属试剂的量显著降低而引起成本有效地使用阴离子链转移催化剂。因此,甲基苯化合物(甲苯)、聚甲基苯化合物(二甲苯、均三甲苯、均四甲苯等)是用于形成适合进一步合成细化的低分子量聚苯乙烯组合物的优异链转移剂。这种甲基苯链转移剂部分地归功于其化学事实的有效性,即它们的最强酸性碳氢键的pka比聚(苯乙烯基)阴离子的共轭酸的pka低至少一个数量级(即,它们酸性更强)。已经报道了更复杂的烷基取代的苯有机链转移剂,特别是最简单的乙苯(eb)(epo741147)或者至少认为其是在使用由叔丁醇钾、仲丁基锂和n,n,n’,n’-四甲基乙二胺(tmeda)形成的催化剂时形成阴离子链转移苯乙烯分布的合适有机链转移剂。对于低分子量组合物,烷基取代的芳烃链转移剂通常构成所述阴离子链转移组合物的相对大百分比的分子量。例如,由甲苯和苯乙烯形成的mw=716的阴离子链转移组合物基于所述组合物的重量平均包含12.8%甲苯。同样,由乙苯和苯乙烯形成的mw=730的阴离子链转移组合物基于所述组合物的重量平均包含14.5%乙苯。可能期望形成低分子量聚苯乙烯组合物,其基本上不含(包含少于组合物的2重量%)的这种有机链转移剂。另外,可能期望阴离子链转移剂(无论是有机的还是无机的)的量也小于所得聚苯乙烯组合物的2重量%。对于阴离子链转移方法,每个和每一个链恰好并入一个添加到有待聚合的单体中的有机链转移引发剂单体。烷基取代的芳烃的分子量≥92.14道尔顿(甲苯的式量)。因此,一个简单的数学事实是,包含≥2重量%的甲苯的阴离子链转移分布的最低mw由mw≥92.14±0.02=4607道尔顿给出。由较高分子量的烷基取代的芳族链转移剂(例如,乙苯或二甲苯)形成的所有阴离子链转移组合物将必须具有大于4607道尔顿的最小mw。理论上,由乙苯和苯乙烯形成的阴离子链转移组合物具有与仅由苯乙烯单体形成的阴离子聚合的苯乙烯组合物相同的结构。然而,恰恰相反,已经发现乙苯在现有技术条件下如epo741147中用作链转移剂时,这种工艺条件提供具有其中添加有杂质和杂质分布的不均匀微结构的聚苯乙烯组合物(参见图12)。这种不期望的杂质和杂质分布来自链异构化和碎裂过程(参见图1和2)。因此,诸如epo741147的实施例的组合物的链长分布全部是包括异构聚合物微结构的不期望集合的分布-这些微结构对芳族亲电取代反应不太理想。这类杂质和杂质分布在这类阴离子链转移组合物的进一步衍生化方面可能是有问题的。就生产市场产品而言,高水平的任何离散杂质-1000ppm或更高的量-是不期望的。因此,期望苯乙烯聚合形成具有大大降低或基本上没有不期望的聚合物微结构特征或杂质片段的低分子量聚苯乙烯组合物,并且期望所述组合物仅包含(/98重量%)的聚合的苯乙烯单体。已经计算出epo741147的阴离子链转移苯乙烯反应分布的乙苯重量%(重量%eb=106/mw*100%)并呈现在下表i中。根据如表i中所呈现的实验细节,通过比较ep0741147实施例2-7可以看出,仅实施例4生成具有有限宽度(标准偏差)和小多分散度的聚合阴离子链转移苯乙烯反应分布(actsr分布)。如所报道的相对进料速率或馈料或两者同时的微小变化导致actsr分布具有非常大的标准偏差并且具有显著增加的多分散度,并且在一些情况下天文数字般地增加(例如,epo741147实施例2、3和7)。因此,从这类实验细节中可以看出,提供用于生成具有窄宽度,即小标准偏差(σn)的分布的非常窄且有限的工艺窗。根据ep0741147a1的实验细节,如下表i中所呈现,通过比较ep0741147实施例2至7和d可以看出,仅实施例4生成具有有限宽度(标准偏差)和小多分散度的阴离子链转移苯乙烯反应分布(actsr分布)。如所报道的相对进料速率或试剂馈料或两者同时的微小变化导致actsr分布具有非常大的标准偏差并且具有显著增加的多分散度,并且在一些实施例中,天文数字般地增加。因此,从这类实验细节可以看出,提供用于生成具有窄宽度,即小标准偏差σn的分布的非常窄且有限的工艺窗。对该现有技术的研究揭示,该工艺技术存在在烃反应介质中不期望地形成低或有限溶解度的催化剂组合物的问题。期望的是具有催化剂体系,该催化剂体系具有显著改善的烃溶解度,其中生成更均匀(如果不是完全均匀的话)的聚合物链长度分布微结构的较低分子量苯乙烯分布的利用率或效率改善。表i:使用乙苯作为链转移剂的现有技术epo741147阴离子链转移苯乙烯聚合如上文在表iep0741147实施例d中所述,基于单金属锂的催化剂体系在催化苯乙烯与乙苯的阴离子链转移聚合方面是无效的,或者至多是非常低效的。更重要的是,已充分证明单金属tmeda丁基锂催化剂形成试剂在乙苯的金属化方面表现出差的区域选择性。在broaddus撰写的一篇论文(broaddus,c.d.,j.org.chem.1970,35,10.)中,报道乙苯的α位氢以仅38%形成苄基锂,其中邻位锂化发生到9%的程度,间位锂化发生到36%的程度并且对位锂化发生到17%的程度。因此,现有技术教导了使乙苯锂化的单金属锂催化剂形成试剂将生成许多不同的聚合物微结构,这些微结构相对于引发乙苯部分是区域异构链长分布。因此,期望具有一种高效的苯乙烯链转移聚合方法,其中需要涉及乙苯(无论乙苯是添加的还是原位形成的)金属化的阴离子链转移步骤的途径减少(如果未被消除的话),但仍然形成期望的阴离子链转移分布链长微结构。一旦实现这样的方法,则将需要直接一步氢化物加成(例如,一步氢锂化反应)到苯乙烯或其他苯乙烯或其他乙烯基芳族单体。这种氢锂化单体将能够有效地引发更多单体的聚合,随后自分子氢链转移。这种方法需要以活性形式一致且重复地重整氢化物催化剂。因此,这种氢介导的类盐氢化物引发的聚合方法将以高收率产生二聚物及更高级的多聚物,并且特征在于催化剂的量从200%降到10,000%的催化剂效率。对该现有技术(结合wo2010065468a1的比较例46至48)的研究揭示,该工艺技术存在在烃反应介质中不期望地形成低或有限溶解度的催化剂组合物的问题。因此,反应馈料的微小变化可导致产物分布的急剧变化,这由催化剂可用性降低引起。epo741147的方法依赖于在长进料时间(6至18小时)内进行的非常慢的相对进料速率,以试图平衡活跃的和无活性的聚合物链。乙苯作为链转移剂的主要问题是如果乙苯的pka不等于聚(苯乙烯基)阴离子的共轭酸的pka近似值,则乙苯的pka具有相同的数量级。期望具有可提供可溶性催化剂组合物的新催化剂和阴离子链转移聚合条件,从而提供以下优点:1)较低分子量的聚苯乙烯分布,mn<930道尔顿,甚至<700道尔顿;2)更经济地使用试剂;3)较短的时期,更有效地使用聚合反应器且其生产率更高。实际上,本发明的氢介导的类盐氢化物引发的工艺技术提供这样的优点。在氢气氛下的苯乙烯聚合对于苯乙烯的齐格勒纳塔(zeiglernatta)聚合(murahashi,s.、nozakura,s.和utsuharay."polymerizationofstyrenewiththeziegler-nattacatalystsinthepresenceofmolecularhydrogen.”bulletinofthechemicalsocietyofjapan196033431)是已知的。另外,至少有一份关于在氢气氛下苯乙烯茂金属聚合的报告(参考文献14:tomotsu,n.、kuramoto,m.、takeuchi,m.和maezawa,h.(1996).metallocenes1996,96,211.(i)chien,jcw.;tomotsu,n.等人,“syndiospecificpolymerizationofstyrene."journalofmolecularcatalysisa:chemical1998128.1167.)。在乙苯的两种聚合化学形成中,提及苯乙烯的氢化产物。因此,utsuhara及其同事报道了可以在氢气存在下获得低分子量的全同立构聚苯乙烯,尽管除此之外,还发现与聚合反应竞争的另一反应,即苯乙烯氢化成乙苯。在两种氢介导苯乙烯聚合的方法:齐格勒纳塔和茂金属催化中,乙苯在动力学上是惰性的并且代表不可恢复的收率损失。deffieux及其同事报道聚(苯乙烯基)锂分布的氢解(50℃h21atm),导致原位形成能够在很大程度上低效地重新引发苯乙烯在100℃下阴离子聚合的氢化锂(ménoret,s.、deffieux,a.和desbois,p.“initiationofretardedstyreneanionicpolymerizationusingcomplexesoflithiumhydridewithorganometalliccompounds.”macromolecules,(2003)36,5988)。deffieux进一步报道:“但是,就增长而言,向苯乙烯中缓慢加入lih会导致不完全的引发。”deffieux报道,在添加所添加的有机金属路易斯酸试剂(n-bu2mg、sec-bu2mg或bumgobt或i-bu3al)的情况下,lih的溶解度和重新启动效率得到改善,但催化效率仅在50%和150%之间。此外,形成的双金属络合物降低了终止速率,并且在50℃下在环己烷和1.1atmh2下,活性或活跃的聚(苯乙烯基)锂物质的半衰期从未络合的聚(苯乙烯基)锂的40分钟大大增加到双金属络合的聚(苯乙烯基)锂的34小时。事实上,他们报道需要50个大气压(约50巴)h2才能将活跃的聚(苯乙烯基)锂物质的半衰期恢复到50分钟。deffieux及其同事教导可溶性氢化锂是苯乙烯聚合的潜在引发剂:“氢化锂,只要它仍然可溶,至少在100℃下甚至在非极性溶剂中是苯乙烯阴离子聚合的潜在引发剂。该引发剂的效率通过与有机金属衍生物络合来改善,这些有机金属衍生物首先确保其溶解性,然后降低苯乙烯的增长速率。当n-bu2mg、sec-bu2mg用作添加剂时,li-h键不是真正的引发位点,聚合在两个金属原子之间的配体交换之后进行。”“在高温下,h2充当苯乙烯阴离子聚合中的链转移剂。然而,为了有效,其在介质中的浓度应该高,以使平衡向金属氢化物的形成移动。这需要高氢工作压力”。然而,deffieux及其同事需要将lih与路易斯酸如二烷基镁试剂、铝烷基和/或烷基铝氢化物络合以溶解lih。这样形成的这类路易斯酸络合的lih试剂一旦用于引发苯乙烯聚合就不能被有效地还原。因此,这类路易斯酸络合的聚(苯乙烯基)锂链不能被有效地还原,它们的还原也不能使高活性或超活性形式的lih引发剂有效地再生。文献中记载,已知仅两种高度可溶的第1族金属氢化物不含路易斯酸络合剂(参见:stasch,a.和fohlmeister,l.aust.j.chem.2015,68,1190-1201.;和liptrot,d.j.,springerthesis:group2mediateddehydrocoupling,第2章group1-group2bimetallicalkylsandhydrides,springerinternationalpublishing,2016,第41至61页)。这些是:(1)“超级聚集体”[(t-buoli)16(lih)17],丁基锂叔丁醇锂的混合物的光解分解产生(thomas,d.等人,j.am.chemsoc.1997,119,11998;和thomas,d.等人,angew.chem.int.ed.1998,37,1537);(2)stash的烃可溶性lih络合物[(dipnpph2)4li8h4](dip,2,6-ipr2c6h3),通过将苯基硅烷应用于反应性金属前体制备(stasch,a.angew.chem.int.ed.2012,51,1930。)然而,该烃可溶性lih试剂不具有反应性或不足以使非常活泼的物质二苯基乙炔或1,1-二苯基乙烯氢锂化。因此,本领域普通技术人员将理解,[(dipnpph2)4li8h4]不太可能使活性甚至更低的苯乙烯单体或其他活性较低的乙烯基芳族单体氢锂化,因此不会引发这类单体的聚合。stash还报道了lih/li(pz)(pz=3,5-二叔丁基-1h-吡唑)的“初始澄清溶液”的形成,其可能由于形成胶体lih而变成乳状。这类“初始澄清溶液”通过在芳族或脂族溶剂中用超过一当量的正丁基锂处理3,5-二叔丁基-1h-吡唑(pzh),然后添加苯基-或二苯基硅烷以将过量的烷基锂基团转化为氢化物来制备。stash通过与lih/li(pz)方法相同的合成策略再次使用空间要求的吡唑特配体(pz)通过[na(pz)]、[na(nbu)]和二苯基硅烷在芳族溶剂中的反应制备了第一种可溶性nah络合物[(pz)6na7h]。应用用以生成[(pz)6na7h]以形成kh氢化物类似物的相同合成策略仅导致结晶聚合物[k(pz)]的形成和分离。因此,迄今未知直接由分子氢h2形成的稳定的脂族和/或脂环族和/或芳族烃可溶性单金属、双金属或多金属碱(第1族)金属氢化物。在他们的出版物中(stasch,a.和fohlmeister,l.aust.j.chem.2015,68,1190-1201)教导以下内容:“定义明确的第1族金属的纯粹氢化物络合物非常罕见,并且实际上目前仅以锂和钠著称...大多数涉及碱金属和氢化氢中心的分离化合物是混合元素体系,并且最佳描述为‘盐’型络合物,其中氢化物配体的最强相互作用是与非碱金属中心或准金属发生的......这使得这些‘盐’络合物中的大多数是共价氢化物络合物。该化合物类型中最突出的实例可能是lialh4、nabh4和其他相关的商业衍生物,如l-selectride®、n-selectride®和k-selectride®(锂、钠、钾三仲丁基(氢化)硼酸盐)或具有空间要求的配体的衍生物。”重点补充。因此,应该清楚的是,现有技术路易斯酸络合的氢化锂、氢化钠和氢化钾引发剂(例如,deffieux和同事在其延迟的苯乙烯阴离子聚合中使用的那些)是共价氢化物,而不是本发明的类盐氢化物催化剂。与共价氢化物形成对比,类盐氢化物(意指离子氢化物)定义为存在作为带负电荷的离子的氢h-与碱金属或碱土金属组合。关于苯乙烯的加成以及不含络合路易斯酸的类盐氢化物的同时聚合,deffieux及其同事提供以下背景(同上):“据我们所知,很少有论文涉及由金属氢化物引发的乙烯基单体的阴离子聚合。williams简要提到了一种在25℃下在己烷中由nah引发的苯乙烯聚合实验。然而,引发效率非常低,并且3天后转化率仅达到90%”。liao及其同事报道具有纳米(约20nm)粒度分布的高活性碱金属氢化物的形式(liao,s.等人,journalofmolecularcatalysis,1993,84,211。)在该论文中,liao报道由相应的碱金属和氢气(1atm)在thf(40℃)中由ticl4和萘催化形成高活性类盐氢化物(hash)。完全转化成类盐氢化物,lih*需要2小时,nah*需要4小时,并且kh*需要28小时(*表示高活性或超活性氢化物)。发现这些纳米类盐氢化物在某些芳基卤化物的脱氯和脱溴中具有一些实用性。据报道,当在某些过渡金属络合物中使用时,它们具有作为烯烃如1-己烯的氢化的助催化剂的活性。报道了在0.003s-1至45.3s-1范围内的转换频率。因此,当结合过渡金属催化剂(1摩尔)使用时,高活性碱金属氢化物(50摩尔至300摩尔)仅还原烯烃,而没有公开烯烃的聚合或甚至二聚化。liao及其同事后来报道了纳米级碱金属氢化物的其他应用(liao,s.等人,synth.comm.1997,273977。)这类应用包括将羰基碳还原为醛和/或苯甲醛的醇、苯甲酸甲酯、丙烯醛和丙烯酸的甲酯和正丁酯。反应在回流的thf中使用化学计量过量的高活性类盐氢化物(以nah*或kah*形式)进行,并且反应时间为0.25小时至15小时。特别感兴趣的是用nah*还原丙烯醛(0.3小时)和丙烯酸甲酯(0.25小时)以分别以97%和96%的收率产生烯丙醇。在另一篇公告中,liao及其同事报道,与cp2ticl2、cp2ticl2-mh(m=li、na或k)络合的热处理的纳米lih、nah和kh可用作使苯乙烯(m=li或na)或辛烯(m=k)氢化的催化剂。纳米kh与cp2ticl2在1大气压h2下并未使苯乙烯氢化,而是引发聚合以形成具有宽熔点范围t=160℃至180℃的非常高分子量(mw)的聚苯乙烯(mw=200,000)。进一步发现,单独的纳米kh使苯乙烯聚合,本领域的普通技术人员将理解,这类高mw阴离子聚苯乙烯(aps)组合物是低效引发并因此导致仅形成很少的活跃聚合物链的结果,这些聚合物链快速地掺入苯乙烯单体中,代价是剩余不溶性纳米kh。zhang及其同事报道在甲苯(9ml)中在氢气氛下在-17℃至42℃下使苯乙烯(2ml)氢化的高活性催化剂(zhang,m.等人,catallett2008,124,146.)。高活性催化剂由纳米尺寸的氢化钠(20mg,8.7×10-4)和12种不同的无路易斯碱的二茂钛络合物(0.5ml的4×10-4摩尔/l,即2×10-7摩尔)–nah*/ti=4350)形成。在其中二茂钛络合物含有配位氧(醚)或氮(叔胺)物质的另外两个实例中未观察到氢的吸收。尽管nah*对于二茂钛催化剂大量过量,但没有报道或甚至提及苯乙烯的聚合,更不用说任何形式的链转移化学。schleyer及其同事报道了超活性(极细分)的锂、钠和钾氢化物形式的制备(schleyer,p.v.r.等人,j.org.chem.1987.52,4299;和schleyer,p.v.r.等人,angewchemint.ed.engl.198625465。)这些超活性类盐氢化物(sash)作为细悬浮液的制备需要在n,n,n’,n’-四甲基乙二胺(tmeda)存在下在己烷中氢化相应的烷基碱金属。超活性lih*的形成在30℃和35℃之间进行,超活性nah*在低温条件(-10℃至-15℃)下制备,并且据报道超活性kh*在-20℃至-25℃下形成。schleyer研究了氢化物在有机合成中的应用,并在上面引用的公开文献中报道。大多数合成反应(金属化、加成和还原)是在低至-90℃的低温条件下进行,很少的反应在室温和50℃之间进行。schleyer也没有公开使用氢化物进行苯乙烯或乙烯基聚合,更不用说这种聚合过程的氢介导。harger及其同事已经报道苯乙烯可用2.5摩尔%的初始由苯基硅烷形成的有机钙催化剂[dippnacnaccah∙thf]2催化氢化(20℃,20个大气压的h2,在苯中15小时)(参见,harder,s.、speilman,j.、buch,f.angew.chem.2008,120,9576,还公布为angew.chem.int.ed.2008,47,9434.)。氢化生成乙苯,收率为85%,还有15%的收率的主要由苯乙烯二聚物和痕量的苯乙烯三聚物和四聚物组成的低聚物。harder还报道了1,1-二苯基乙烯在由5摩尔%丁基锂/tmeda络合物形成的催化剂中在20℃、20大气压h2下在苯中15小时以低转化率还原,以产生14%ph2chch3和7%的二聚物。关于该反应,作者作出以下声明:“由商业上可获得的nbuli/tmeda催化的反应仅进行到低转化...这提出,在较低的h2压力下,较重的碱土金属络合物是更有效的催化剂”。ashby及其同事由邻位取代的(2,6-二甲基-和2,6-二异丙基苯氧化物)芳氧基镁试剂和活性形式的固体氢化镁生成四氢呋喃可溶形式的氢化镁。四氢呋喃不溶形式的氢化镁由烷氧基镁试剂和固体氢化镁试剂产生(参见,ashbey,e.c.、goel,a.b.、lin,j.j.tetrahedronletters,1977,3133。)ashby还报道了通过与活性形式的固体氢化镁反应由一系列本体二烷基和本体烷基取代的环烷基仲胺形成四氢呋喃可溶性二烷基氨基镁氢化物。所述活性形式的氢化镁通过用lialh4在乙醚中还原二甲基镁制备。因此,本体的二烷基和本体的烷基取代的环烷基仲胺与二甲基镁反应以形成双(二烷基镁)镁化合物,其又在thf中与活性形式的氢化镁反应(参见ashbey,e.c.、goel,a.b.、lin,j.j.j.or.chem.,1978,43,1564。如果这类氨基镁氢化物可以引发聚合,则其可能将通过向单体中添加酰胺而在一定程度上引发聚合,并导致不期望的胺官能团并入所得聚合物分布中。michalczyk报道了在“适当的配体”存在下在醚或烃溶剂中形成沉淀形式的氢化镁mgh2lx。这类适当的配体包括四氢呋喃、乙二醇二甲醚和tmeda。所用的还原剂为苯基硅烷(参见michalczyk,m.j.organometallics,1992,11,2307)。在最近一篇题为“molecularearlymaingroupmetalhydrides:syntheticchallenge,structuresandapplications”的综述中,harder回顾了明确定义的第1族和第2族金属氢化物的受控合成的最新技术。通常,这类氢化物通过上面概述的方法制备,这些方法包括:光降解;活性氢化物反应形成“ate-络合物”,诸如芳氧基镁氢化物以及由ashby报道的二烷基氨基镁氢化物;harder的[dippnacnaccah∙thf]2初始由苯基硅烷形成;以及stasch的可溶性氢化锂络合物,由苯基硅烷形成。另外,harder回顾了由菱镁矿复合物[(ipr2n)3mg-]m+(m+=na+、k+)的热分解形成的大量氢化物。形成可溶性类盐氢化物组合物的所有方法的共同特征是使用本体(通常是异丙基化的配体)来实现溶解性。在所有情况下,除了在苯乙烯氢化成乙苯(85%收率)期间形成的催化性差的物质,例如使用[dippnacnaccah∙thf]2(其同样初始由苯基硅烷形成)的实例外,类盐氢化物络合物由除分子氢以外的其他一些试剂形成。只有scheyer不溶形式的超活性类盐氢化物(sash)直接由分子氢作为初始还原剂形成。因此,现有技术没有公开锂氨基醇盐络合的类盐氢化物(loxsh)物质用于乙烯基芳族单体如苯乙烯类单体的阴离子链转移聚合的用途。事实上,现有技术甚至没有预料到loxsh催化剂,尤其是作为烃可溶性物质的形成,并且特别是在由简单的非本体配体形成时,更不用说是直接由h2形成。本发明人已经发现这些氢化物以及以下令人惊讶的事实:使用这些新型氢化物可以催化氢介导的类盐氢化物引发的聚合过程。因此,本发明提供一种在温和温度(例如,约20℃至低于100℃)下使乙烯基芳族单体有效阴离子链转移聚合的方法,其中氢是主要或唯一的链转移剂。所述方法可以在相对低至非常低的氢分压下进行。此外,本发明人已经发现本发明的新型聚合催化剂提供仅由苯乙烯(/98重量%苯乙烯)组成的低分子量阴离子聚合的苯乙烯分布,具有独特、均匀且有益的“头对尾”微结构,基本上(如果不是全部)没有聚合物微结构的季碳。因此,这类聚苯乙烯分布具有小于3.0重量%,优选小于2.0重量%,更优选小于1.0重量%的聚合物链骨架中具有季碳的聚苯乙烯聚合物链。因此,这类组合物全部具有小于1000ppm,甚至小于200ppm且甚至小于20ppm的存在的季碳。同样地,本发明的聚苯乙烯组合物具有小于1.0%,优选小于0.5%且最优选小于0.1%的由期望的阴离子链转移聚苯乙烯分布的断裂产生的聚合物链分布或杂质。化学缩略语和数字术语的定义pta是用作促进剂的一般类型的聚叔胺的首字母缩写,使用“•xpta”,其中x是正数并且表示与催化剂组合物络合的pta的摩尔数,为整数或分数。tmeda是n,n,n’,n’-四甲基乙二胺pta的首字母缩写,使用“•xtmeda”,其中x是正数并且表示使用和/或与催化剂组合物络合的tmeda的摩尔数,为整数或分数。pcah是用于形成本发明催化剂的一般类型的极化络合剂的首字母缩写,表示极化络合剂作为中性醇,使用[pca‾]表示极化络合剂为已经将一个质子让给更碱性的化学物质的醇盐的形式。因此,下文中使用[pca‾]是为了方便地显示醇盐已由pcah形成。使用诸如[pca‾]xmyhz的式应解释为中性催化剂络合物或聚集体,其中y个金属原子的电荷由x个[pca‾]阴离子的电荷与z个氢化物离子组合平衡。应该理解,使用[pca‾]xmyhz是针对整个催化剂制剂而言,并且将包含任何潜在的“盐(ate)’络合物,诸如:[[pca‾]xmyhz-n]n+[[pca‾]xmyhz+n]n‾。dmeah是作为中性氨基醇的n,n-二甲基乙醇胺的首字母缩写(同义词:n,n-二甲基-2-羟基乙胺、n,n-二甲基乙醇胺),使用[dmea‾]表示n,n-二甲基乙醇胺为已经将一个质子让给更碱性的物质的醇盐的形式。dmaeoeh是作为中性氨基醚-醇的2-n,n-二甲基氨基乙氧基乙醇(n(ch3)2ch2ch2o-ch2ch2oh)的首字母缩写,使用[dmaeoe‾]表示n,n-二甲基氨基乙氧基乙醇为已经将一个质子让给更碱性的物质的醇盐的形式。meoeh是作为中性醚-醇的2-甲氧基乙醇的首字母缩写,使用[meoe‾]表示2-甲氧基乙醇为已经将一个质子让给更碱性的物质的醇盐的形式。阴离子链转移方法的效率(effct)由以下表达给出:effct.=mnth/mnexp;其中mnth是理论数均分子量,并且术语mnexp是在实际运行或方法中获得的数均分子量。效率百分比通过效率乘以100%获得。用于描述分子量分布的参数的简短摘要和定义它们的公式呈现在下表ii中。(a.rudin,theelementsofpolymerscienceandengineering,academicpress,orlando,1982,第54至58页)。数均dp(dpn)使用mn作为100%聚苯乙烯组合物计算。表ii参数公式dpn,数均聚合度dpn=(mn-2)/104(对于聚苯乙烯分布)mn,数均分子量mn=(σmini)mw,重均分子量mw=[(σmi²ni)/mn]mz,z均分子量mz=(σmi³ni)/σmi²nipd,多分散指数(也称pdi)pd=(σmini)/[(σmi²ni)/mn]方差v=(mwmn-mn²)标准偏差,σnσn=√(mwmn-mn²)skewness,nu3nu3=mzmwmn-3mn²mw+2mn³不对称性,nα3nα3=(mzmwmn-3mn²mw+2mn³)/σn³技术实现要素:本发明的氢介导的类盐氢化物引发的聚合(hmship)方法的特征在于:a)可溶性类盐氢化物物质快速加成到乙烯基芳族单体上以形成引发物质的新能力;b)发生类盐氢化物物质加成到单体上且因此允许重新引发步骤与增长反应步骤之间竞争以生长活性瞬时的活跃聚(苯乙烯基)阴离子链,从而维持恒定或接近恒定数量的活性生长链方面的新的高效率;和c)在温和且新型的工艺条件下自氢转移链以终止这类活跃的聚(苯乙烯基)阴离子物质,并且再生呈能够有效且高效地重新引发聚合过程的形式的类盐氢化物的能力。因此,本发明涉及一种进行氢介导的类盐氢化物引发的聚合的方法,该方法的特征在于在包含分子氢的气氛下将一种或多种阴离子可聚合的烃单体进料到含有可溶性类盐氢化物催化剂的反应介质中。在没有这些特征的情况下,该化学方法将以其他方式在一个极端上主要生成还原的单体或者在另一个极端生成高分子量聚合物。在本发明的一些实施方案中,前述特征协同作用并平衡竞争,产生具有高收率、高效率和优异控制的聚合物链长微结构的阴离子链转移聚合物分布。本发明还涉及一种用于阴离子链转移聚合的方法,其包括在反应器容器中在包含分子氢的气氛下将乙烯基芳族单体和/或优选苯乙烯类单体进料到反应混合物中,其中所述反应混合物由以下各物形成:(i)有机锂化合物和/或有机镁化合物;(ii)任选地,聚叔胺化合物;(iii)选自叔氨基醇化合物、叔氨基醚-醇、醚-醇或其组合的极化络合剂;(iv)任选地,碱金属或金属合金和/或固体类盐氢化物和/或类盐金属酰胺;(v)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;(vi)任选地,乙烯基芳族单体;和在(vii)pka大于h2的烃溶剂中;其中芳烃和烃溶剂可以相同或不同;(viii)任选地,分子氢;并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。本发明还涉及一种用于阴离子链转移聚合的方法,其包括在具有式[dmea‾]xliyhz的氢介导的链转移聚合催化剂的反应器容器中在包含分子氢的气氛下将乙烯基芳族单体和/或优选苯乙烯类单体进料到反应混合物中,其中所述催化剂由以下物质的接触过程形成:(i)约y当量的有机锂化合物和/或有机镁化合物;(ii)任选地,tmeda化合物;(iii)约x当量的二甲基氨基乙醇;(iv)任选地,乙苯;(v)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;和(vi)分子氢,其中形成的氢化物的量z由公式z=y-x给出,并且x、y和z是大于零的正实整数或分数;其中所述式可以进一步包含n,n,n’,n’-四甲基乙二胺(tmeda)配体络合物,即[dmea‾]xliyhz•xtmeda,摩尔比为x摩尔tmeda/摩尔催化剂[dmea‾]xliyhz,其中x=0.0001至约8.0。因此,本发明还涉及一种用于阴离子链转移聚合的方法,其包括:在反应器容器中在包含分子氢的气氛下(a)进料苯乙烯单体;和/或(b)共进料苯乙烯单体和反应混合物,其中所述反应混合物初始由以下各物形成:(i)约y当量的有机锂化合物;(ii)任选地,tmeda化合物;(iii)约x当量的二甲基氨基乙醇;(iv)任选地,乙苯;和(v)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;(vi)任选地,分子氢,其中形成式[dmea‾]xliyhz的烃可溶性氢化锂,其中形成的氢化物的量z由公式z=y-x给出,并且x、y和z是大于零的正实整数或分数;并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。本发明还涉及高度烃可溶性双金属叔氨基醇盐和/或叔氨基醚-醇盐和/或醚-醇盐络合的氢化锂和/或氢化镁催化剂和/或试剂,其由包含以下各物的反应介质形成:(i)分子氢;(ii)有机锂化合物和/或有机镁化合物;(iii)任选地,聚叔胺化合物;(iv)选自叔氨基醇化合物、叔氨基醚-醇、醚-醇或其组合的极化络合剂;(v)任选地,碱金属或金属合金和/或固体类盐氢化物和/或类盐金属酰胺;(vi)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;(vii)任选地,乙烯基芳族单体;和(viii)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。本发明还涉及高度烃可溶性单金属叔氨基醇盐和/或叔氨基醚-醇盐和/或醚-醇盐络合的氢化锂或多氢化锂(lihn,其中n=1+2x,其中x是正整数)络合物和/或聚集体,其由包含以下各物的反应介质形成:(i)分子氢;(ii)有机锂化合物;(iii)任选地,聚叔胺化合物;(iv)选自叔氨基醇化合物、叔氨基醚-醇、醚-醇或其组合的极化络合剂;(v)任选地,锂金属或锂合金和/或固体氢化锂和/或氨基锂;(vi)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;(vii)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。本发明还涉及高度烃可溶性锂叔氨基醇盐络合的氢化锂(lih)或多氢化锂(lihn,其中n=1+2x,其中x是正整数)氢介导的链转移聚合催化剂,其具有式[dmea‾]xliyhz,其中所述催化剂由使以下物质接触的过程形成:(i)约y当量的有机锂化合物和/或有机镁化合物;(ii)任选地,tmeda化合物;(iii)约x当量的二甲基氨基乙醇;(iv)任选地,乙苯;(v)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;和(vi)分子氢,其中形成的氢化物的量z由公式z=y-x给出,并且x、y和z是大于零的正实整数或分数;其中所述式可以进一步包含n,n,n’,n’-四甲基乙二胺(tmeda)配体络合物,即[dmea‾]xliyhz•xtmeda,摩尔比为x摩尔tmeda/摩尔催化剂[dmea‾]xliyhz,其中x=0.0001至约8.0。本发明还涉及形成的高度烃可溶性叔氨基醇盐和/或叔氨基醚-醇盐和/或醚-醇盐络合的氘化锂(li2h)、或氚化锂(li3h)或多氘化锂(li2hn,其中n=1+2x,其中x是正整数)或多氚化锂(li3hn,其中n=1+2x,其中x是正整数)络合物和/或由包含以下各物的反应介质形成:(i)同位素富集的分子氢;(ii)有机锂化合物和/或有机镁化合物;(iii)任选地,聚叔胺化合物;(iv)选自叔氨基醇化合物、叔氨基醚-醇、醚-醇或其组合的极化络合剂;(v)任选地,碱金属或金属合金和/或固体类盐氢化物和/或类盐金属酰胺;(vi)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;(vii)任选地,乙烯基芳族单体;和(viii)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。本发明还涉及高度烃可溶性锂叔氨基醇盐络合的氢化锂(lih)或多氢化锂(lihn,其中n=1+2j,其中j是包括零的正整数)络合物和/或聚集体,其由包含以下各物的反应介质形成:(i)分子氢;(ii)约y当量的有机锂化合物;(iii)任选地,tmeda;(iv)约x当量的二甲基氨基乙醇;(vi)任选地,乙苯;和(vii)pka大于h2的烃溶剂;其中乙苯和烃溶剂可以相同或不同;其中形成式[dmea‾]xliyhn的烃可溶性氢化锂,其中形成的氢化物的量z由公式z=y-x给出,并且x、y和z是大于零的正实整数并且n=z+2j;并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。本发明还涉及氢介导的苯乙烯阴离子聚合(hmaps)方法,其特征在于:a)可溶性氢化锂物质使苯乙烯快速聚合的新能力;b)发生氢化锂物质加成到苯乙烯单体上且因此允许重新引发步骤与增长反应步骤竞争以生长活性瞬时的活跃聚(苯乙烯基)锂阴离子链,从而维持恒定或接近恒定数量的活性生长链方面的新的高效率;c)在温和和新型工艺条件下从氢转移链以终止这类活跃聚(苯乙烯基)锂阴离子物质并且再生呈能够有效重新引发聚合过程的形式的可溶性氢化锂的能力;d)消除或几乎消除分子内链转移步骤,否则会导致在阴离子聚苯乙烯聚合物链中形成不期望的季碳;和f)mwd的控制通过高相对单体与催化剂进料速率、催化剂浓度和氢活性来实现。因此,本发明还涉及一种用于阴离子链转移聚合的hmaps方法,其包括在反应器容器中在包含h2的气氛下将苯乙烯单体进料到反应混合物中,其中所述反应混合物含有具有化学式[dmea‾]xliyhz的催化剂,其中所述催化剂由使以下物质接触的过程形成:(i)约y当量的有机锂化合物和/或有机镁化合物;(ii)任选地,tmeda化合物;(iii)约x当量的二甲基氨基乙醇;(iv)任选地,乙苯;(v)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;和(vi)任选地,分子氢;其中形成的氢化物的量z由公式z=y-x给出,并且x、y和z是大于零的正实整数或分数并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。本发明还涉及类盐氢化物引发的阴离子聚苯乙烯和阴离子链转移聚苯乙烯分布,其中各种或离散聚合物链成分的聚苯乙烯链长分布具有以下通式:其中n是在引发苯乙烯单体和终止苯乙烯单体之间共价键合的重复苯乙烯单体单元的数目;其中上述通式的链长分布占聚苯乙烯组合物总重量的至少约97.0重量%,优选至少约98.0重量%,更优选至少约99.0重量%且最优选占至少约99.2重量%;并且其中链长分布的余量占不超过3.0重量%,或更优选不超过2.0重量%,更优选不超过1.0%,且最优选占不超过0.8重量%,该链长分布的余量是以下的组合:1)1的异构体的聚合物链的分布;和/或2)由链断裂工艺步骤形成且因此基本上不含具有由以下公式给出的结构式量(fwi)的离散链长组合物的分布:fwi=fwi聚苯乙烯±14道尔顿。本发明进一步涉及氢化锂引发的阴离子聚苯乙烯和阴离子链转移聚苯乙烯分布,其中各种或离散聚合物链成分的聚苯乙烯链长分布具有通式1:其中n是在引发苯乙烯单体和终止苯乙烯单体之间共价键合的重复苯乙烯单体单元的数目;其中上述通式的链长分布占聚苯乙烯组合物总重量的至少约99.0重量%且优选占至少约99.2重量%;并且其中链长分布的余量占不超过1.0重量%,或更优选不超过0.8重量%,该链长分布的余量是以下的组合:1)式1的异构体的聚合物链的分布;和2)基本上不含由链断裂工艺步骤形成的链长分布且因此具有离散链长结构式量(fwi),其中fwi=fwi聚苯乙烯±14道尔顿,这低于由gc低聚物测试确定的约0.02%的检测极限。实施方式词汇应理解和体会到,如本文中包括权利要求的任何地方使用的术语“聚合物”是指如“聚合物”的oecd定义的上下文中定义的术语“聚合物”,其如下:“化学物质由以一种或多种类型的单体单元的序列为特征的分子组成,并且包含多数简单重量的含有至少三个单体单元的分子,这些单体单元与至少一个其他单体单元或其他反应物共价键合并且由少于多数简单重量的相同分子量的分子组成。这类分子必须分布在一定分子量范围内,其中分子量的差异主要归因于单体单元数量的差异”。类盐氢化物(意指离子氢化物)定义为存在作为带负电荷的离子h‾的氢与碱金属或碱土金属组合,所述碱金属包括锂、钠、钾、铷和铯;并且所述碱土金属包括镁和钙。类盐金属酰胺是由氨和/或伯胺和/或仲胺形成的金属酰胺或二酰胺,其中与酰胺组合的金属离子是选自锂、钠、钾、铷和铯的碱金属;并且与二酰胺组合的是金属离子是碱土金属并且包括镁和钙。这里使用的“聚合物微结构”是指就组成、序列分布、立体构型、几何和取代异构而言的离散聚合物链(或这类链的链长分布)构型。术语“头对尾”聚合物微结构是由上面呈现的苯乙烯聚合物结构(1)表现出的微结构的描述。当每个苯乙烯类单体单元的头部(由带有苯基取代基的乙烯基碳形成)共价键合到一个且仅一个其他苯乙烯类单体单元的尾部(由亚乙烯基碳形成)时,存在头对尾微结构。当苯乙烯类单体单元的尾部共价键合到另一苯乙烯类单体单元的尾部时,存在术语“尾对尾”微结构。这类尾对尾微结构是通过电子转移机制引发的苯乙烯类单体的阴离子聚合的微结构的常见的一部分。术语“尾对头对尾”微结构是指聚合物主链连接,其中一个苯乙烯类单体的头部共价键合到另外两个苯乙烯类单体的尾部。该微结构在聚合物主链中生成不规则性,并且在聚合物链中并入不期望的化学不稳定(在某些反应条件下易于裂解)的季碳。术语“有机锂(活性)烷基”、“活性”锂和“活性有机锂烷基”(缩写为li活性)和术语“有机镁(活性)烷基”和“活性有机镁烷基”是指作为高于滴定任何质子试剂以及任何质子杂质物质如水和/或醇和/或伯胺或仲胺所需的有机锂和/或有机镁化合物的量馈入的金属烷基的这些有机金属化合物中任一种的总量。尽管我们希望不受理论束缚,但认为活性有机锂的摩尔量等于基于1:1形成的类盐氢化物的摩尔量。还认为1摩尔当量的活性有机镁化合物形成最多2当量摩尔的类盐氢化物。为此目的,“活性金属烷基”表示锂和/或镁共价键合的烷基基团,其中键合的烷基基团可以是脂族、脂环族、芳族、烯丙基、苄基或乙烯基烃基。质子在与术语物质或试剂或溶剂或杂质组合时是指在本发明的化学工艺的条件下具有pka低于h2的共价键合的质子(h+)的化学物质(参见buncel,e.、menon,bj.am.chem.soc.,1977,99,4457:“carbanionmechanisms.6.metalationofarylmethanesbypotassiumhydride/18-crown-6etherintetrahydrofuranandtheacidityofhydrogen”)。“loxsh”是指由以下物质形成的锂氨基醇盐或锂胺-醚-醇盐或锂-醚-醇盐络合的类盐氢化物;(i)分子氢;(ii)有机锂化合物,有或没有有机镁化合物;(iii)任选地,聚叔胺化合物;(iv)叔氨基醇和/或叔氨基醚-醇和/或醚-醇;(v)任选的固体碱金属或碱土金属氢化物或碱金属或碱金属合金;(vi)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;和在(vii)pka大于h2的烃溶剂中;其中芳烃和烃溶剂可以相同或不同(参见:daasbjerg,k,actachemicascandinavica,1995,49,878:“estimationofthepkaforsomehydrocarbonsandaldehydesandsolvantionenergiesofthecorrespondinganions”)。loxlih是表示单金属形式的loxsh的术语,其中催化剂/试剂用锂试剂作为唯一的金属试剂形成。loxkh是表示由锂和钾组成的双金属催化剂的术语,其中活性类盐氢化物的一部分是氢化钾。loxmgh2是表示由锂和镁组成的双金属催化剂的术语,其中活性类盐氢化物的一部分是氢化镁。[dmea‾]xliyhz表示由以下物质形成的烃可溶性锂氨基醇盐络合的氢化锂的催化剂或试剂组分的化学式:(i)分子氢;(ii)约y当量的有机锂化合物;(iii)任选地,tmeda;(iv)约x当量的二甲基氨基乙醇;(v)任选地,乙苯;和在(vii)pka大于h2的烃溶剂中;其中烃溶剂可以是乙苯或不同(参见:daasbjerg,k,actachemicascandinavica,1995,49,878:“estimationofthepkaforsomehydrocarbonsandaldehydesandsolvantionenergiesofthecorrespondinganions”);其中指数值x、y和z是正实数,其中形成的氢化物的当量是z,其中z=y-x,并且其中,对于化学计量比x:y:z;a)y在约2至约6的范围内;:b)x在约1至约5的范围内;并且因此z在约5至约1的范围内。[dmea‾]xliyhz的优选组合物是催化剂组合物,其中化学计量比x:y:z为2:3:1[dmea‾]2li3h或其中[dmea‾]4li6h2或[dmea‾]6li9h3或者x=2,y=3且z=1的任何更高的数字倍数。术语“分子氢”是指h2为1h2,但也可以包括氢2h2或3h2的同位素,无论是作为同位素的混合物还是富含特定的同位素,无论是在蒸气空间中的气态还是溶解在凝相中。术语“碱金属合金”是指至少两种金属的金属合金,其中至少一种金属是碱金属,然而这一碱金属合金可以由两种碱金属组成,诸如nak或nak2,并且可以具有溶解的这类碱金属或以具有该合金的一些物理组合。术语“和/或”表示单一的或组合的。例如,“a和/或b”表示单独的“a”、单独的“b”或a与b的组合。术语“有或没有”表示单一的或组合的。例如,有或没有b的a表示单独的“a”或a与b的组合。术语“约x当量”、“约y当量”、“约z当量”等表示自化学计量当量的±50%,或±30%,或±20%,或±10%,或±5%,具有以下规定:量x总是小于活性y(即“活性有机锂试剂,又称活性锂)的总量,因此量z是大于但不等于零的正实数。术语“有机锂化合物”是指键合到锂原子的有机基团。有机基团的非限制性实例可以是脂族(例如,烷基基团)、脂环族(例如,环烷基)、乙烯基基团、烯丙基、苄基、芳族基团(例如,苯基)或聚(苯乙烯基)锂。术语“有机镁化合物”是指键合到镁原子的有机基团。有机基团的非限制性实例可以是脂族(例如,烷基基团)、脂环族(例如,环烷基)、乙烯基基团、烯丙基、苄基、芳族基团(例如,苯基)或聚(苯乙烯基)镁。优选的有机镁化合物是具有两个有机基团的有机镁化合物。“术语聚叔胺(pta)促进剂”是指含有至少两个叔胺基团的化合物,其在hmship过程期间促进或活化氢化物催化剂的形成。非限制性通式将包括:其中r'和r”独立地是能够与两个或多个胺形成键的有机基团,并且r1、r2、r3、r4和r5独立地是有机基团,它们也可以进一步被其他叔胺取代,并且指数值n独立地是等于或大于0的整数(即n=0、1、2、3......)。应当理解,当n=0时,括号内的基团不存在,并且该结构旨在表示化学键位于与括号的两侧相交的两个基团之间。因此,当n=0时,聚叔胺结构2变为结构4。术语“极化络合剂”是用于形成本发明催化剂的中性醇的总称,所述中性醇诸如叔氨基醇、叔氨基醚-醇或醚-醇。术语“碱金属或碱土金属醇盐”、“碱金属或碱土金属醚-醇盐”和“碱金属或碱土金属醚-醇盐”分别是由叔氨基醇或叔氨基醚-醇或醚-醇和碱金属和/或碱金属或碱土金属氢化物,和/或碱金属或碱土金属酰胺或和/或碱金属或碱土金属烷基形成的醇盐。叔氨基醇或叔氨基醚-醇或醚-醇可由(但不限于)以下通用结构表示:其中r是能够与一个或多个叔胺和一个羟基形成键的有机基团,r1独立地是有机基团,其也可以进一步被其他叔胺取代,σ可以包括:i)对于5、6、7、8、9和10,o或nr1;和ii)对于11,o或nr1或ch2;并且指数值n独立地是等于或大于0的整数(即n=0、1、2、3......)。应当理解,当n=0时,括号内的基团不存在,并且该结构旨在表示化学键位于互连括号的两侧的两个基团或原子之间。优选的氨基醇包括二甲基氨基乙醇、二乙基氨基乙醇、3-二甲基氨基-1-丙醇、n-甲基-二乙醇胺、三乙醇胺、2-[2-(二甲基氨基)乙氧基]乙醇、1-(2-羟乙基)哌啶、1-(2-羟乙基)吗啉、1-(2-羟乙基)吡咯烷、1-甲基-2-吡咯烷甲醇等等。优选叔氨基醇或叔氨基醚-醇或醚-醇不经历除了形成醇盐的反应之外的另外金属化反应。因此,并非所有的叔氨基醇或叔氨基醚-醇或醚-醇都适用于形成一些催化剂组合物,尤其是用过量的某些有机锂化合物-特别是烷基锂试剂形成的组合物。过量是指摩尔量大于用于形成催化剂的叔氨基醇或叔氨基醚-醇或醚-醇的醇部分的摩尔量。另外,叔氨基醇盐或叔氨基醚-醇盐或醚-醇盐应当用于增溶旁观配体。这意味着除了起到活化极化络合剂的作用外,它还赋予催化剂组合物的类盐氢化物溶解性并有助于在hmship过程期间活化和形成类盐氢化物。因此,极化配体另外是惰性的,并且不参与聚合过程,也不参与催化剂降解反应。不期望将这类配体的叔氨基醇或叔氨基醚-醇或醚-醇降解产物或并入聚合物结构或产物分布中。附图说明图1是说明导致活跃的(意指离子化)阴离子苯乙烯三聚物或苯乙烯三聚物等效物(即,乙苯与两种苯乙烯单体组合)异构化和进一步聚合以形成和不期望的季化“尾对头对尾”微结构的化学反应途径的图。图2是说明化学反应途径的图,所述化学反应途径需要异构化的活跃(意指离子化或作为聚(苯乙烯基)阴离子或烷基)阴离子苯乙烯三聚物或苯乙烯三聚物等价物的断裂,随后聚合以形成具有由以下公式给出的式量的聚合物链:fwi=[i(104)+2–14]道尔顿以及具有fwi=[i(104)+2+14]道尔顿的离散低聚物。图3是具有所需高纯度“头对尾”苯乙烯低聚物的结构归属(structuralassignment)的气相色谱图,这类苯乙烯低聚物是采用本发明的氢介导的类盐氢化物引发的聚合方法用由本发明的loxlih催化剂形成的本发明的聚苯乙烯组合物获得。图4是苯乙烯低聚物的气相色谱图,这类苯乙烯低聚物是采用本发明的氢介导的类盐氢化物引发的聚合方法用由本发明的loxlih催化剂形成的本发明的聚苯乙烯组合物获得。图5是具有苯乙烯低聚物的结构归属的气相色谱图,这类苯乙烯低聚物是采用本发明的氢介导的类盐氢化物引发的聚合方法用由hash催化剂形成的聚苯乙烯组合物获得。图6是具有苯乙烯低聚物的结构归属的气相色谱图,这类苯乙烯低聚物是采用本发明的氢介导的类盐氢化物引发的聚合方法用由loxkh催化剂形成的聚苯乙烯组合物获得。图7是具有苯乙烯低聚物的结构归属的气相色谱图,这类苯乙烯低聚物是采用本发明的氢介导的类盐氢化物引发的聚合方法用由sash催化剂形成的聚苯乙烯组合物获得。图8是具有痕量的由未还原的丁基镁引发的“头对尾”苯乙烯低聚物以及高纯度的“头对尾”苯乙烯低聚物的结构归属的气相色谱图,这类高纯度的“头对尾”苯乙烯低聚物是采用本发明的氢介导的类盐氢化物引发的聚合方法用由本发明的loxmgh2催化剂形成的本发明的聚苯乙烯组合物获得。图9是具有提供为用于比较的标准物的在氮气氛下由正丙基苯和苯乙烯形成的wo2008154453的比较actvap组合物的结构归属的气相色谱图。图10是具有在氮气氛下由甲苯和苯乙烯形成的wo2008154453的比较actsp组合物的结构归属的气相色谱图,该比较actsp组合物提供为用于比较的标准物以鉴定由不期望的断裂聚合过程形成的fwi=[i(104)+2-14](其中i=2-6)道尔顿。图11是具有α-甲基苯乙烯与乙苯的比较单加合物的结构归属的气相色谱图,该比较单加合物提供为用于比较的标准物以鉴定由导致原位形成α-甲基苯乙烯的后续并入的不期望的断裂聚合过程形成的fwi=[i(104)+2+14](其中i=2)道尔顿。图12是由epo741147的苯乙烯工艺技术的乙苯链转移聚合获得的低聚物的气相色谱图,证明由该技术的催化剂和方法的特征性异构化和断裂聚合反应产生的不期望水平的聚合物微结构。图13是由wo2008154453的苯乙烯(1摩尔份)工艺技术的乙苯(2摩尔份)链转移聚合获得的低聚物的气相色谱图,证明当乙苯为链转移剂时由该技术的催化剂和方法的特征性异构化和断裂聚合反应产生的不期望水平的聚合物微结构。图14是由wo2008154453的苯乙烯(2摩尔份)工艺技术的乙苯(1摩尔份)链转移聚合获得的低聚物的气相色谱图,证明当乙苯为链转移剂时由该技术的催化剂和方法的特征性异构化和断裂聚合反应产生的不期望水平的聚合物微结构。图15是苯乙烯低聚物的气相色谱图,该苯乙烯低聚物是采用在约80℃下进行的本发明的氢介导的阴离子苯乙烯聚合(hmaps)方法用由本发明的另一种loxlih催化剂[dmea‾]xliyhz•2tmeda(其中x:y:z为约3:2:1)形成的本发明的聚苯乙烯组合物获得,证明99.94%的头对尾微结构。图16是苯乙烯低聚物的气相色谱图,该苯乙烯低聚物是采用在约80℃下进行的本发明的氢介导的阴离子苯乙烯聚合(hmaps)方法用由本发明的另一种loxlih催化剂[dmea‾]xliyhz(其中x:y:z为约3:2:1)形成的本发明的聚苯乙烯组合物获得,证明99.97%的头对尾微结构。图17是苯乙烯低聚物的气相色谱图,该苯乙烯低聚物是采用在约90℃下进行的本发明的氢介导的阴离子苯乙烯聚合(hmaps)方法用由本发明的另一种loxlih催化剂[dmea‾]xliyhz(其中x:y:z为约3:2:1)形成的本发明的聚苯乙烯组合物获得,证明99.93%的头对尾微结构。具体实施方式描述本发明涉及一种进行阴离子可聚合的烃单体的氢介导的类盐氢化物引发的聚合(hmship)的方法,用于进行这一方法的催化剂组合物以及在某些优选的条件下形成非常纯的“头对尾”微结构的新型且有益的低分子量阴离子链转移聚合物分布。该方法的特征在于在包含分子氢的气氛下将至少一种阴离子可聚合的烃单体进料到含有活性且通常可溶的类盐氢化物催化剂的合适溶剂中,其中自分子氢的链转移是决定动力学链长(υ)分布且因此所得产物分布的数均分子量(mn)的机理的重要分量。本发明的一个实施方案涉及一种使用烃可溶性loxsh催化剂进行乙烯基芳族单体如苯乙烯类单体如苯乙烯的氢介导的阴离子聚合的方法。该烃可溶性loxsh催化剂由包含以下各物的反应介质形成:(i)有机锂化合物,有或没有有机镁化合物;(ii)任选的聚叔胺促进剂化合物;(iii)选自叔氨基醇、叔氨基醚-醇、醚-醇或其组合的极化络合剂;(iv)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;(v)任选的固体碱金属或碱土金属氢化物或碱金属或碱金属合金或碱金属或碱土金属酰胺;(vi)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;和(vii)分子氢。当单体仅为苯乙烯时,由这一方法形成的产物分布在下文中被称为loxshps分布。更具体地讲,当催化剂是仅由锂作为金属组成的单金属催化剂时,该催化剂被称为loxlih,并且所得阴离子链转移苯乙烯聚合物分布为loxlihps。然而,当催化剂是由锂和钾组成的双金属催化剂时,该催化剂被称为loxkh,并且所得阴离子链转移苯乙烯聚合物分布为loxkhps。同样,当催化剂是由锂和镁组成的双金属催化剂时,该催化剂被称为loxmgh2,并且所得阴离子链转移苯乙烯聚合物分布为loxmgh2ps。在本发明的实践中,loxsh催化剂可以任选地以多种方法形成,这些方法不受限制但包括:a.在惰性气氛下在(vi)中形成由(i)、任选的(ii)、(iii)、任选的(iv)和任选的(v)组成的充分混合并反应的溶液或悬浮液,然后通过以下方法转化为loxsh:1)将一部分单体进料到由此形成的“盐”络合物中;然后2)用h2替换或以其他方式置换惰性气氛;或者b.在惰性气氛下在(vi)中形成由(i)、任选的(ii)、(iii)、任选的(iv)和任选的(v)组成的充分混合并反应的溶液或悬浮液,以形成前体“盐”络合物,然后通过用氢气替换或以其他方式置换惰性气氛将其转化为loxsh;或者c.在外部反应器中在(vi)中形成由一部分的(i)以及任选的(ii)、(iii)、期望量的(iv)和任选的(v)组成的充分混合并反应的溶液或悬浮液,在氢气下将所述溶液转移到聚合反应器中,然后馈入余量的(i);或者d.在氢气氛下在(vi)中形成由任选的(ii)、(iii)、任选的(iv)和任选的(v)组成的充分混合并反应的溶液或悬浮液;进料一部分单体,然后一次性进料(i);或者e.在氢气氛下在(vi)中形成由任选的(ii)、(iii)、任选的(iv)和任选的(v)组成的充分混合并反应的溶液或悬浮液,然后在大于约3分钟的时间内进料(i),然后任选地或在必要时进料单体;或者f.在氢气氛下在(vi)中形成由(iii)、任选的(iv)和任选的(v)组成的充分混合并反应的溶液或悬浮液,然后在大于约3分钟的时间内进料(i),接着添加期望量的(ii),然后任选地或在必要时进料单体。已经发现loxlih和loxmgh2催化剂/试剂根据方法e和f在1.0至2.0个大气压h2压力的氢气氛下以非常活跃且可溶的形式方便地制备,尽管可以采用更高或更低的h2压力。然而,loxmgh2催化剂没有被还原成氢化物或者至少没有被氢气完全还原成氢化物,直到引入单体苯乙烯。苯乙烯的引入形成聚(苯乙烯基)镁试剂,其在氢介导的链转移聚合期间被完全还原成氢化物。因此,对于loxmgh2,可能需要存在乙烯基芳族单体以在进料单体是必需的而不是任选的情况下完全形成催化剂。如下面实施例中所述,在形成本发明的loxlih和loxmgh2催化剂和或试剂时,采用约-5℃至约40℃的初始温度。特别是在用氨基醇盐时,可以使用更低或更高的初始温度来形成催化剂。因此,然后使用在计量针阀上的适度背压将有机锂(正丁基锂)和或有机镁(二正丁基镁)化合物(i)缓慢进料到包含在(vi)中的溶解的h2、(ii)、(iii)的充分搅拌的反应混合物中。如果有机锂和/或有机镁化合物在催化剂形成过程期间产生轻质烃如丁烷,则最初热启动随即发生并且反应器压力将升高。随后观察到温度继续升高,然而,反应器压力通常会降至初始压力以下或者随着在催化剂形成过程中消耗氢气而保持恒定。当初始压力为0psig时,在反应器中产生或可以产生负压或轻微真空。在大多数情况下,在实施例中所述的条件下,在有机锂试剂的进料期间h2压力通常会下降。然而,在一些实施例中,当温度低于15℃时和或当使用有机镁试剂时,在引入有机金属试剂期间反应器压力似乎不下降。而是在将反应混合物加热至所需反应温度的同时发生还原成氢化物。在loxmgh2催化剂的情况下,氢吸收似乎并不显著,直至在70℃或更高的反应温度下引入单体苯乙烯。从loxmgh2运行获得的最低分子量低聚物的质谱分析证明存在痕量水平的低聚物,其已经用丁基基团引发;在任何其他不含镁的催化剂体系中都没有观察到这种情况。为了在形成催化剂反应混合物时促进loxsh催化剂的还原,将氢气进一步馈入反应器中,使得一旦将反应混合物加热到所需的初始聚合反应温度,反应器压力就将为40psig至70psig,尽管可以并且已经采用更高的压力。在形成本发明的催化剂组合物:loxsh;loxlih;loxmgh2;或可通过本发明生成的试剂时,可使用0.1巴至大于300巴的氢气压力。优选的h2压力在0.25巴至10.00巴范围内,更优选的范围为0.5巴至7.0巴,且最优选的范围为0.75巴至约5.0巴。我们没有忘记的是,高达100gpa的压力在理论上能够或者实际上可能由本发明的催化剂和试剂中生成多氢化物组合物,因此这类压力的应用和这类产物的形成都在本发明的范围内。应注意的是,可商购获得的有机镁化合物可含有约0.12重量%至0.25重量%的作为试剂的添加剂的三乙基铝(tea),其通常在庚烷中供应。因为这类有机铝试剂可以对阴离子聚合反应具有延迟作用,所以期望锂金属与铝金属的比率大于50.0:1.0,且优选大于101.0:1.0。如图8中所绘,没有证据表明tea引发或参与loxmgh2运行的hmship过程。因此,通过类推,其他有机金属试剂如有机铝和或有机硼鎓和或有机硼试剂可以存在于反应混合物中,只要添加的试剂不会延迟hmship过程到抑制氢化物加成单体上和/或阻止或以其他方式明显妨碍氢链转移到增长的聚合物链上并因此形成不期望的分子量参数的分布即可。适合形成loxsh催化剂的有机锂化合物的非限制性优选实例是正丁基锂、仲丁基锂、叔丁基锂、烯丙基锂、乙烯基锂、苯基锂、1-己基-1-苯基锂、1-己基-1,1-二苯基锂、环己基锂和聚(苯乙烯基)锂化合物,它们可以是添加的或原位产生的。适合形成loxmgh2催化剂的有机镁化合物的非限制性优选实例是丁基乙基镁(bem)、二正丁基镁(dbm)、正丁基正辛基镁、二正辛基镁、二环己基镁和聚(苯乙烯基)镁化合物。潜在的有机镁化合物的综合列表提供在美国专利3,817,955中。在形成loxmgh2催化剂时,可能预先形成具有化学计量r3mgli或r4mgli2的“盐”络合物,其中基团r独立地是烷基、乙烯基、环烷基、聚(苯乙烯基)、苯基,其选自但不限于有机锂与上述有机镁的任何组合。在形成loxsh催化剂的过程中形成的非限制性碱金属或碱土金属氨基醇盐、碱金属或碱土金属氨基醚-醇盐和碱金属或碱土金属醚-醇盐(对于碱金属和碱土金属醇盐分别称为[pca-]m+或[pca-]2m2+)由上文合适的极化络合剂[pcah]的通用结构形成。应该清楚的是,形成的[pca-]m+和/或[pca-]2m2+可以在形成loxsh催化剂时原位形成和/或它们可以提前很好地形成并作为碱金属或碱土金属氨基醇盐、碱金属或碱土金属氨基醚-醇盐和碱金属或碱土金属醚-醇盐馈入催化剂形成反应器。因此任何能够由pcah和/或[pca-]前体(例如,适当还原的适当的n,n-二烷基氨基酸)形成[pca-]m+和/或[pca-]2m2+的碱金属或碱土金属试剂可用于实施本发明,因此在本发明的范围内。如实施例25至27中所证明,催化剂组合物的[pca-]组分可以提前形成并随后在催化剂形成反应期间馈入。因此,诸如固体碱和碱金属氢化物、碱金属和碱金属合金、碱金属和碱土金属烷基、碱金属和/或碱土金属酰胺(类盐金属酰胺)的试剂可用于实施本发明以与pcah和/或[pca-]前体反应以形成[pca-],该[pca-]包含本发明的催化剂和试剂。所述[pca-]m+和/或[pca-]2m2+的形成也可以如下进行:(a)在催化剂形成之前和/或催化剂形成期间在催化剂形成反应器中原位进行;和/或(b)在与催化剂形成反应器相关或与催化剂形成反应器完全分离的外部反应器中进行。然而,应该注意的是,使用类盐金属酰胺可能潜在地导致胺官能团并入本发明的聚合物组合物中,并且在一些应用中是不期望的。另外,使用络合的金属氢化物如liah4以形成[pca-]li+可能需要将如此形成的[pca-]li+与这一反应的铝共产物分离。下文的实施例28和29证明,氨基醚-醇盐可在某些条件下在形成时或在loxlih催化的hmship过程期间降解或分解。实施例30证明,在某些工艺条件下,用由2-甲氧基乙醇和有机锂化合物形成的醚-醇盐形成的loxlih催化剂不能充分地活化氢链转移过程。因此,特别用于loxlih方法的优选的极化络合剂是氨基醇盐。然而,应该理解,氨基醚-醇盐可以很好地适用于低lih活性的loxsh双金属催化剂。同样地,通过实施本发明可以发现由2-甲氧基乙醇形成的loxlih催化剂更合适或更具活性的条件或方法。有许多可用的手性叔氨基醇,这些手性叔氨基醇可以通过例如手性氨基酸的还原和进一步合成精制来合成,因此合适氨基醇的列表可以是无穷无尽的。使用这类手性叔氨基醇可以提供在选择性、立构规整度或立体规择性方面的任何优势都在本发明的范围内。尽管如此,可以通过胺与反应性环醚如环氧乙烷或其他环氧化物的简单反应制备的不太复杂的叔氨基醇是优选的。易得的这种叔氨基醇的非限制性实例包括:二甲基氨基乙醇、二乙基氨基乙醇、n-甲基-二乙醇胺、3-二甲基氨基-1-丙醇、2-[2-(二甲基氨基)乙氧基]乙醇、1-(2-羟基乙基)哌啶、1-(2-羟乙基)吗啉、1-(2-羟乙基)吡咯烷、1-甲基-2-吡咯烷甲醇等。可用于loxsh催化剂的聚(叔胺)促进剂的非限制性实例包括衍生自丙二胺的二(叔胺)配体、衍生自乙二胺或衍生自聚乙烯亚胺的二(叔胺)配体。优选的实例包括n,n,n’,n’-四甲基乙二胺(tmeda)、n,n,n′,n′′,n′′-五甲基二亚乙基三胺(pmdeta)、金雀花碱(sparteine)、鹰爪豆碱(isosparteine)和1,4-甲基哌嗪,其中tmeda是最优选的。最优选的聚(叔胺)促进剂配体是最易挥发和/或最大水和/或酸溶性的化合物,因此tmeda是优选的。聚叔胺促进剂化合物的存在似乎有助于形成loxsh催化剂/试剂。loxsh方法可以在不存在聚叔胺促进剂的情况下进行,然而在一些实施例中聚叔胺促进剂的存在提供较低不对称性的loxshps分布,并且在降低的单体进料速度下且在降低的氢气压力下以增加的收率提供该loxshps分布。可以使用的优选的芳烃是相对于甲苯具有±2.75pka单位的任何芳烃,然而,可以想到的是可以采用芳烃如具有比甲苯低的4.32单位的pka的二苯基甲烷,只要:1)在聚合物微结构中并入二苯基甲烷部分不影响最终用途;和/或2)在反应条件下,这类烃的pka充分高于h2的pka,以便不妨碍氢介导的链转移机理。可以使用的芳烃的非限制性实例是苯、甲苯、均三甲苯、乙苯、正丙基苯、正丁基苯、异丁基苯、戊基苯、1,2-二芳基乙烷、1,3-二芳基丙烷、枯烯、叔丁基苯、1-烷基萘、2-烷基萘或苯乙烯二聚物或低分子量低聚物分布(苯乙烯二聚物、三聚物、四聚物和五聚物)。虽然这类芳烃的使用是任选的,但是它们的使用是优选的,因为据信它们的存在减少或抢占或以其他方式减轻有机锂,更具体地烷基锂试剂对聚叔胺促进剂的不期望的攻击。loxsh催化剂可以减轻或抑制有机锂试剂对包含所述催化剂的碱金属或碱土金属氨基醇盐、碱金属或碱土金属氨基醚-醇盐和“碱金属或碱土金属醚-盐的攻击。优选通过蒸馏或通过聚合物沉淀从产物分布中容易地除去的烃。用于使用苯乙烯的hmship方法的最优选的芳烃是乙苯。可使用的烃溶剂是在反应条件下具有大于分子氢(h2)的pka的任何烃。这类优选溶剂的非限制性实例是环己烷、甲基环己烷和上面列出的芳烃。可以使用其他烃溶剂,只要它们的使用不影响类盐氢化物催化剂、活性中间体、瞬时活跃的聚合物链和聚合物链分布产物的溶解度。芳烃和芳族溶剂可以相同或不同。这意味着芳烃可以充当芳烃和溶剂两者。例如,乙苯是苯乙烯聚合中的优选组分,并且可以用作芳烃和溶剂两者。在这种情况下,对于loxsh过程,组分(iv)和(vi)将合并成一种组分(或限制)并且是相同的。同样,它们可以是不同的。例如,芳烃可以是乙苯,并且烃可以是环己烷。因此,组分(iv)和(vi)将是不同的。此外,如果不使用芳烃并且例如,将环己烷用作组分(vi),则组分(iv)可以是任选的。上述loxsh催化剂形成过程中氢的分压维持在约0.001巴至约10.0巴,或约0.3巴至约6.8巴,或约0.5巴至约5.2巴或约1.0巴至约4.2巴的压力下。在由叔氨基醇和有机锂化合物形成单金属loxlih催化剂时,叔氨基醇与有机锂的比率可以广泛变化。然而,应该理解,为了形成氢化锂物质,必须使用相对于摩尔当量的叔氨基醇摩尔过量的有机锂化合物,使得锂-碳键可用于还原得到氢化锂物质。可以设想采用每41摩尔的有机锂试剂40摩尔的叔氨基醇的馈料比;然而,这种馈料比是对昂贵试剂的浪费。优选的馈料比(叔氨基醇:有机锂)在(1.00:1.05)至约(1:6)的范围内,更优选的范围是(1.00:1.10)至约(1:5);甚至更优选的范围是(1.0:1.2)至约(1:4);并且最优选的范围是(1.00:1.40)至约(1:3)。尽管我们希望不受理论束缚,但下面实施例33和34中所述的结果表明n,n-二甲基氨基乙醇(dmeah)与正丁基锂的(1:1.5)-即(2:3)馈料比,同时或随后用氢还原形成氢化物,得到具有式[dmea‾]4li6h2的二氢化物催化剂,其中两种氢化物中只有一种在环己烷气氛下引发苯乙烯的聚合。类似地,实施例35和36表明1:2比率的dmeah与正丁基锂提供具有式[dmea‾]4li8h4的四氢化物催化剂,其中四种氢化物中的仅一种在氢气氛下引发苯乙烯的聚合。同样,如实施例37)中所用的(1:3)馈料比生成具有式[dmea‾]2li6h4的可溶性氢化物催化剂,其中仅约1/9的氢化物在环己烷气氛中引发苯乙烯的聚合。因此,理论上该优选的催化剂组合物包括具有经验式[dmea‾]4li12h8与[dmea‾]5li15h10组合的聚集体的混合物,其中两种催化剂仅一种氢化物可用于在没有另外分子氢的情况下引发苯乙烯聚合。最优选的催化剂组合物由具有以下总经验式的催化剂聚集体组成:(a)[dmea‾]5li12h7;和/或(b)[dmea‾]2li5h3,其中(叔氨基醇:有机锂)的比率在(1:2.4)至(1:2.5)的范围内。相同的优选范围适用于由叔氨基醚-醇和/或醚-醇形成可溶性氢化锂络合物、催化剂和试剂。应该注意的是,据信当在氢气下形成时,loxlih催化剂作为聚集体存在,其在某些化学计量下作为固定分子量的明确定义的物质存在,而其他化学计量或馈料比提供未明确定义但作为非均匀聚集体的混合物存在的催化剂。进一步相信,聚叔胺促进剂的存在可另外稳定某些聚集体或有助于将较大聚集体的其他不太均匀的混合物分解成更小的更具活性的聚集体。因此,催化剂体系的活性可以在很大程度上变化,但总体上这类催化剂在引发活跃阴离子聚合方面相对较差。也就是说,作为引发剂,相对于用于引发的氢化物的可用性而言,在无h2气氛中的loxlih试剂相对低效(effct<1.0并且在effct=0.1至effct=0.67的范围内)。因此,令人格外惊讶的是这些相同的催化剂如此有效地引发和催化本发明的氢介导的阴离子链转移聚合,其中%effct=1000%至16000%,较低的值(接近1000%的值)仅由单体的有意限制使用产生。烃可溶性loxsh催化剂可具有以下经验化学式:a)[pca‾]4li6h2;b)[pca‾]4li8h4;c)[pca‾]2li6h4;d)[pca‾]4li12h8;e)[pca‾]5li15h10;f)[pca‾]5li12h7;g)[pca‾]2li5h3;h)[pca‾]4li4mgh2;i)[pca‾]4li4mg2h4;j)[pca‾]2li4mgh4;k)[pca‾]4li6mg3h8;l)[pca‾]5li9mg3h10;m)[pca‾]5li6mg3h7;n)[pca‾]2li3mgh3;o)[pca‾]4li5kh2;p)[pca‾]4li7kh4;和q)[pca‾]2li5kh4,并且其中所述经验式可任选地进一步包含pta促进剂配体络合物,其中总碱金属和碱土金属与pta的摩尔比为约10,000:1.0至约1.0:约8.0。优选的[pca‾]是[dmea‾]、[dmaeoe‾]或[meoe‾]。在形成双金属第i族碱金属loxsh催化剂时,叔氨基醇与总碱金属的比率(叔氨基醇:碱)在(1.00:1.05)至约(1:6)的范围内,更优选的范围为(1.00:1.10)至约(1:5);甚至更优选的范围为(1.0:1.2)至约(1:4);并且最优选的范围为(1.00:1.40)至约(1:3)。并且锂与除锂外的碱金属的比率(li:na和/或li:k和/或li:cs等)为(10,000:1)至(1:2),优选的比率在(34:1)至(2:1)范围内,且最优选为(17:1)至(3:1)。应该理解的是,在这方面,(10,000:1)的馈料比可以表示由于在反应器或馈料线或罐中从有意馈入碱金属的先前运行留下的量而导致意外存在甚至痕量的碱金属,特别是钾。基于钾和其他碱金属的双金属锂催化剂具有在反应器壁上沉积痕量水平的催化剂或催化剂副产物的趋势,已经发现这类痕量水平在氢介导的类盐氢化物引发苯乙烯聚合期间负面影响由纯loxlih催化剂体系递送的另外高选择性微结构。可由本发明生成具有以下经验化学式的双金属锂与钠和/或与钾和/或与铯的双金属络合物loxsh催化剂:a)[dmea‾]4na4li4h4;b)[dmea‾]6na4li4h2);c)[dmea‾]6na4li6h4;d)[dmea‾]4k4li4h4;e)[dmea‾]6k4li4h2);f)[dmea‾]6k4li6h4;g)[dmea‾]4cs4li4h4;h)[dmea‾]6cs4li4h2);和i)[dmea‾]6cs4li6h4。另外,这类双金属loxsh催化剂制剂与上述单金属loxlih催化剂制剂组合在本发明的范围内。在用锂形成基于氢化镁的双金属第ii族碱土金属络合物loxmgh2催化剂时,其中锂提供1当量且镁提供2的(叔氨基醇:总金属当量)的比率在(1.00:1.05)至约(1:6)范围内,更优选的范围为(1.00:1.10)至约(1:5);甚至更优选的范围为(1.0:1.2)至约(1:4);并且最优选的范围为(1.00:1.40)至约(1:3)。因此,具有经验式[dmea‾]4li4mgh2(4摩尔dmeah:5摩尔总金属量)的催化剂组合物因此是(4:6或1.0:1.5dmeah:总金属当量)并且是优选的。同样,具有经验式:a)[dmea‾]4li4mg2h4(1:2dmeah:总金属当量);b)[dmea‾]6li4mg2h2(1.00:1.33dmeah:总金属当量);和c)[dmea‾]6li4mg3h4(1.00:1.67dmeah:总金属当量)的催化剂;单独或与上述loxlih或loxsh催化剂制剂组合在本发明的范围内。可以使用在由叔氨基醚-醇和/或醚-醇形成可溶性双金属锂氢化锂络合物、催化剂和试剂中相同的优选范围。可以应用相同的优选范围以由叔氨基醇和/或叔氨基醚-醇和/或醚-醇形成可溶性双金属锂钙氢化物络合物、催化剂和试剂。本发明进一步涉及一种由包括固体碱金属氢化物、碱金属和/或碱金属合金的试剂形成的烃可溶性催化剂或试剂组合物,其中极化络合剂与总碱金属的比例在约1:1.05至约1:6的范围内;并且有机锂化合物与碱金属的摩尔比为10,000:1至1:2。另一个实施方案是由分子氢和以下物质形成的烃可溶性单金属loxlih催化剂或试剂组合物:a.极化络合剂反应形成锂醇盐和有机锂化合物,其中极化络合剂与总碱金属的摩尔比在约1:1.05至约1:6的范围内;或者b.极化络合剂和有机锂化合物,其中极化络合剂与总碱金属的摩尔比在约1:1.05至约1:6的范围内;或者c.极化络合剂;固体氢化锂和/或锂金属的至少一种;和有机锂化合物,其中极化络合剂与总锂的摩尔比在约1:1.05至约1:6的范围内。另一个实施方案是烃可溶性催化剂或试剂组合物,其包含由分子氢和以下物质形成的氢化镁碱土金属loxmgh2催化剂或试剂:a.极化络合剂反应以形成锂和/或镁醇盐;有机锂化合物和/或有机镁化合物;其中极化络合剂与总金属当量的摩尔比在约1:1.05至约1:6的范围内,其中锂提供1当量且镁提供2当量;或者b.极化络合剂;有机锂化合物;和有机镁化合物;其中极化络合剂与总金属当量的摩尔比在约1:1.05至约1:6的范围内,其中锂提供1当量且镁提供2当量;或者c.极化络合剂;固体氢化锂、金属锂、固体氢化镁中的至少一种;有机锂化合物和/或有机镁化合物中的至少一种;其中极化络合剂与总金属当量的摩尔比在约1:1.05至约1:6的范围内,其中锂提供1当量且镁提供2当量;或者d.极化络合剂;固体氢化锂和/或金属锂中的至少一种;和有机镁化合物;其中极化络合剂与总金属当量的摩尔比在约1:1.05至约1:6的范围内,其中锂提供1当量且镁提供2当量;e.极化络合剂;固体氢化镁;和有机锂化合物;其中极化络合剂与总金属当量的摩尔比在约1:1.05至约1:6的范围内,其中锂提供1当量且镁提供2当量;f.并且其中催化剂组合物中锂与镁的摩尔比为10,000:1至1:6。另一个实施方案是用于由以下物质形成烃可溶性催化剂或试剂组合物的方法:(i)分子氢;(ii)有机锂化合物和/或有机镁化合物;(iii)任选地,聚叔胺化合物;(iv)选自叔氨基醇化合物、叔氨基醚-醇、醚-醇或其组合的极化络合剂;(v)任选地,碱金属或金属合金和/或固体类盐氢化物和/或碱金属酰胺;(vi)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;和在(vii)pka大于h2的烃溶剂中;其中芳烃和烃溶剂可以相同或不同。在上述方法中,用于形成催化剂或试剂的氢分压在约0.1巴至约300巴的范围内。此外,用于形成催化剂或试剂的温度在约-96℃至约130℃范围内。此外,极化络合剂与有机锂化合物和/或有机镁的摩尔当量馈料比在每摩尔的金属-烷基当量约1:1.05至约1:6摩尔的极化络合剂范围内,其中锂提供1当量且镁提供2当量。当pta用于形成催化剂或试剂时,pta与总金属(镁和锂)的摩尔比为约10,000:1.0至约1:8。形成烃可溶性催化剂或试剂组合物的一个实施方案包括以下步骤:a.首先使极化络合剂与以下至少一种醇盐形成试剂接触:a)固体氢化锂;b)锂金属;c)氢化镁;d)氨基锂;e)氨基镁;f)有机锂化合物;g)有机镁化合物,由此形成反应混合物,其中极化络合剂与醇盐形成试剂的化学计量摩尔当量比为约1:1至小于1:1,其中锂提供1当量,且镁提供2当量;b.其中进一步添加有机锂化合物和/或有机镁化合物;c.其中极化络合剂与总金属当量的比率在约1:1.05至约1:6的范围内,其中锂提供1当量,且镁提供2当量;d.其中锂与镁的摩尔比在约10,000:1至约1.0:6.0的范围内;e.并且其中所形成的反应产物用氢气进一步还原,以形成烃可溶性类盐氢化物催化剂或试剂。形成烃可溶性催化剂或试剂组合物的另一个实施方案包括以下步骤:a.首先使极化络合剂与碱金属醇盐形成试剂接触,从而形成反应混合物,其中极化络合剂与醇盐形成试剂的化学计量摩尔当量比为约1:1至小于1:1;b.其中所述醇盐形成试剂是以下各物中的至少一种:a)固体碱金属氢化物;b)碱金属;c)碱金属合金;d)和碱金属酰胺;e)氨基镁;f)有机锂化合物;g)有机镁化合物;c.其中进一步添加有机锂和/或有机镁化合物;d.其中极化络合剂与总碱金属的比率在约1:1.05至约1:6的范围内;e.其中锂与非锂碱金属的摩尔比在1:2至约10,000:1的范围内,或催化剂组合物的碱金属仅地为锂;f.并且其中所形成的反应产物用氢气进一步还原,以形成烃可溶性类盐氢化物催化剂或试剂。在上述形成烃可溶性催化剂或试剂组合物的方法和步骤中,该方法还可包括进料苯乙烯类单体以形成聚(苯乙烯基)镁和/或聚(苯乙烯基)锂化合物,然后通过与分子氢接触进行还原以形成可溶性类盐氢化物,其中苯乙烯类单体与总金属的摩尔比为约1:10至约20:1。在另一个实施方案中,该方法可以进一步包括进料苯乙烯类单体以形成瞬时1-苯基-己基镁化合物和/或聚(苯乙烯基)镁化合物,其进一步被分子氢还原成可溶性类盐氢化物,其中苯乙烯类单体与镁的摩尔比为约1:5至约20:1。优选的苯乙烯类单体是苯乙烯。在本发明的另一个实施方案中,烃可溶性催化剂或试剂组合物由以下物质形成:(i)分子氢;(ii)有机锂化合物和/或有机镁化合物;(iii)任选地,聚叔胺化合物;(iv)选自叔氨基醇化合物、叔氨基醚-醇、醚-醇或其组合的极化络合剂;(v)任选地,碱金属或金属合金和/或固体类盐氢化物和/或碱金属酰胺;(vi)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;(vii)乙烯基芳族单体;和在(viii)pka大于h2的烃溶剂中;其中芳烃和烃溶剂可以相同或不同;并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。本发明的另一个实施方案是一种由以下物质形成的催化剂或试剂组合物:(i)分子氢;(ii)有机锂化合物和/或有机镁化合物;(iii)任选地,聚叔胺化合物;(iv)叔氨基醇化合物和/或叔氨基醚-醇和/或醚-醇;(v)任选地,碱金属或金属合金和/或固体类盐氢化物;(vi)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;和在(vii)任选地,pka大于h2的烃溶剂中;其中芳烃和烃溶剂可以相同或不同。最期望的loxsh催化剂是当叔氨基醇是n,n-二甲基氨基醇(dmeah)时并且取决于试剂的馈料比形成的双金属loxmgh2催化剂组合物或聚集体,其可具有如下经验和/或分子式作为催化剂混合物在溶液中的明确定义的组成或平均组成:a)[dmea‾]4li4mgh2;b)[dmea‾]4li4mg2h4;c)[dmea‾]2li4mgh4;d)[dmea‾]4li6mg3h8;e)[dmea‾]5li9mg3h10;(f)[dmea‾]5li6mg3h7;和(b)[dmea‾]2li3mgh3。最期望的loxsh催化剂是当叔氨基醇是n,n-二甲基氨基醇(dmeah)时并且取决于试剂的馈料比形成的单金属loxlih催化剂组合物或聚集体,其可具有如下经验和/或分子式作为催化剂混合物在溶液中的明确定义的组成或平均组成:a)[dmea‾]4li6h2;b)[dmea‾]4li8h4;c)[dmea‾]2li6h4;d)[dmea‾]4li12h8;e)[dmea‾]5li15h10;(f)[dmea‾]5li12h7;和(g)[dmea‾]2li5h3。因此,优选的烃可溶性loxlih催化剂具有化学式[dmea‾]xliyhz;其中z=y-x且x、y和z为大于且不等于零的正实整数或分数,并且其中所述式可任选地进一步包含tmeda配体络合物,其总锂与tmeda的摩尔比为约10,000:1.0至约1.0:约8.0。因此,本发明还涉及一种由包括氢、有机锂化合物和二甲基氨基乙醇的试剂形成并取决于试剂的馈料比的烃可溶性催化剂或试剂组合物,其可以具有以下一种或多种的经验和/或分子式作为:i)1)-105)在溶液中的明确定义的lih聚集体组成;或ii)任何比例的1)-105)中的任何两种或更多种在溶液中的平均lih聚集体组成;或iii)在溶液中的一种或多种明确定义的lih聚集体组成或平均组成与不溶解的1)-105)的一些不溶性lih聚集体组成组合:或iv)i)、ii)或iii)中的任一种,其中催化剂聚集体的总组成可表示为[dmea‾]xliyhz。因此,本发明的烃可溶性[dmea‾]xliyhz催化剂由以下物质形成:(i)y当量的有机锂化合物;(ii)任选量的tmeda;(iii)x当量的二甲基氨基乙醇;(iv)任选地,乙苯;在(v)pka大于h2的烃溶剂中,其中芳烃可为乙苯或者不同;和(vi)分子氢;其中形成的氢化物的量z由公式z=y-x给出,并且x、y和z是大于且不等于零的正实整数或分数。在本发明的实践中,[dmea‾]xliyhz催化剂可任选地以各种方法形成,这些方法不限于但包括:a)在惰性气氛下形成由约y当量的(i)、任选的(ii)、约x当量(iii)、任选的(iv)和在(v)中的充分混合并反应的溶液或悬浮液,然后通过以下转化成[dmea‾]xliyhz:1)将一部分的苯乙烯进料到由此形成的“盐”络合物中;然后2)用h2替换或以其他方式置换惰性气氛;或者b)在惰性气氛下在(v)中形成由约y当量的(i)、任选的(ii)、约x当量的(iii)和任选的(iv)组成的充分混合并反应的溶液或悬浮液,以形成前体“盐”络合物,然后通过用氢气替换或以其他方式置换惰性气氛将其转化成[dmea‾]xliyhz;或者c)在外部反应器中在(vi)中形成由一部分的约x当量的(i)以及任选的(ii)、约x当量的(iii)和任选的(iv)组成的充分混合并反应的溶液或悬浮液,在氢气下将所述溶液转移到氢化物形成反应器中,然后馈入余量的约z或y-x当量的(i);或者d)在氢气氛下在(v)中形成由任选的(ii)、约x当量的(iii)和任选的(iv)组成的充分混合并反应的溶液或悬浮液;然后一次性或以大比例增量进料y当量的(i);或者e)在氢气氛下在(v)中形成由任选的(ii)、约x当量的(iii)和任选的(iv)组成的充分混合并反应的溶液或悬浮液,然后连续地或以增量经大于约3分钟的时间进料约y当量的(i);或者f)在氢气氛下在(v)中形成由约x当量的(iii)和任选的(iv)组成的充分混合并反应的溶液或悬浮液,然后连续地或以增量经大于约3分钟的时间进料约y当量的(i),接着添加期望量的(ii);g)在氢气氛下在一部分的(v)中形成由x当量的(iii)和任选的(iv)组成的充分混合并反应的溶液或悬浮液,然后连续地或以增量经大于约3分钟的时间进料先前用(iv)和/或一部分(v)进一步稀释的约y当量的(i),然后添加期望量的(ii);h)在氢气氛下在一部分的(v)中形成由约x当量的(i)和任选的(iv)组成的充分混合并反应的溶液或悬浮液,然后一次性或连续地或以增量随时间进料先前用(iv)和/或一部分(v)进一步稀释的约x当量的(iii),直至(iii)的全部馈料完成,然后进料约z或y-x当量的(i),接着添加期望量的(ii)。已经发现[dmea‾]xliyhz催化剂/试剂根据方法e)、f)和g)在1.0-3.0大气压的h2压力下方便地以非常活泼且可溶的形式制备,尽管可以采用更高或更低的h2的压力。在反应条件下根据(iv)和(v)的熔点以及(i)、(ii)和(iii)的溶解度和稳定性在形成本发明的[dmea‾]xliyhz催化剂和或试剂时可以采用-96℃至高于100℃的初始温度。如实施例中所述,[dmea‾]xliyhz催化剂可以在35℃至40℃范围的温度下方便地制备。因此,当形成[dmea‾]2li3h催化剂时,最通常将约3当量(y≈3)的有机锂化合物(在环己烷中16.5重量%的正丁基锂)(i)用环己烷和/或甲基环己烷(v)进一步稀释和/或然后在35℃至40℃下将乙苯(iv)缓慢进料(经15分钟至25分钟的时间,使用计量针阀上的适度背压(阀门上的20至30psig背压)到包含在有或没有乙苯(iv)的环己烷和或甲基环己烷(v)中的溶解的h2(15-22psigh2)、约2当量(x≈2)的二甲基氨基乙醇(ii)的充分搅拌的反应混合物(在使用下文描述的节距叶片叶轮或任何其他合适的气体分散混合装置时,约50rpm至约2000rpm;优选约200rpm至约1500rpm;且最优选约500rpm至约1200rpm)。最初在前1/3至约2/3的(i)进料的期间,几乎没有可辨别的热冲击随即发生,并且由于形成的丁烷-即(i)是丁基锂试剂,反应器压力将增加2-3psig。反应器压力的增加也部分地是由引入的进料体积引起的顶空蒸气和气体压缩的结果-特别是在(i)用大量的(iv)和/或(v)进一步稀释时。在最后约1/3的(i)进料期间,温度将升高(在实施例中描述的反应器中,1.0°至3.0°)并且压力将根据用于形成[dmea‾]xliyhz催化剂的条件稳定或降低1psig至3psig。为了在形成催化剂反应混合物时促进[dmea‾]xliyhz催化剂的完全还原,将氢气进一步馈入反应器中,使得一旦将反应混合物加热至所需初始聚合反应温度,反应器压力将为60psig至80psig。已经发现,虽然不必形成活性催化剂,但是当采用该进一步氢气馈料至高压时,制备最具活性形式的[dmea‾]xliyhz催化剂。还发现,当将[dmea‾]xliyhz催化剂保持在所需或接近氢介导的阴离子聚合反应温度(约68℃至约82℃)持续大于约1小时,优选大于约2小时至大于约5小时的时间,然后可以将其放空至所需h2压力以进行聚合时,形成最具活性且最可重现的催化剂。本发明的实施不必执行这类催化剂老化程序;然而,该技术更有利于运行之间的可重现性。尽管我们不希望受理论束缚,但催化剂老化过程可提供最高浓度的可用氢化物,其以在离散聚集体内具有2、3或4个lih部分(取决于(i):(iii)的馈料比)的明确定义的催化剂组合物的形式。催化剂老化过程被认为是每个聚集体具有大于4个lih部分或当量的初始形成的较高级聚集体的平衡或再分布和或低于形成较高浓度的x:y:z的期望比率的催化剂聚集体的x:y:z的期望比率的催化剂聚集体组合物的再分布。因此,在以下情况下形成最具活性的催化剂:(a)将约3当量的(i)(在环己烷中2.0m,1重量份)进一步溶解在8至10重量份的乙苯中;(b)在约35℃至40℃下在16psig至21psig的氢气氛下经约15分钟至25分钟的时间将(a)进料到以约500rpm搅拌溶解在24至28重量份不含(ii)的(iv)和/或(v)中的约2当量的(iii)中;(c)任选地添加(ii);(d)将氢气压力增加至50psig,并将混合rpms增加至900rpm至约1200rpm;(e)将(b)中形成的溶液加热至约65℃至约85℃;(f)将氢气压力进一步增加至约75psig;和(g)将催化剂老化约1小时至大于约5小时的时间。在老化期后,小心地将氢气压力从反应器放空达到期望的压力,以便进行本发明的氢介导的苯乙烯阴离子聚合方法。迄今未知直接由氢形成的不含任何共价键合的路易斯酸基团的烃可溶性氢化锂组合物。因此,在高温下在氢气下的这种平衡或催化剂老化过程也是本发明的特征。根据关于邻位锂氨基醇盐聚集体以及锂氨基醇盐络合的有机锂试剂的形成的公开文献报道,该特征并非没有一些合理性。因此,据报道,由手性邻位锂氨基醇盐制备的某些有机锂试剂通过遵循某些馈料方案并在引入酮之前在低温条件下老化试剂组分,使得有机锂试剂加成到酮上更具对映选择性。在这方面,参见:collumn,d.b.等人,“highlyenantioselective1,2additionoflithiumacetylide-ephedratecomplexes:spectroscopicevidenceforreactionproceedingvia2:2tetramer,andx-raycharacterizationofrelatedcomplexes”,j.am.chem.soc.2000,122.11212。适合形成loxsh催化剂的有机锂化合物的非限制性优选实例是正丁基锂、仲丁基锂、叔丁基锂、烯丙基锂、乙烯基锂、苯基锂、1-己基-1-苯基锂、1-己基-1,1-二苯基锂、环己基锂和聚(苯乙烯基)锂化合物,它们可以是添加的或原位产生的。在由二甲基氨基乙醇和有机锂化合物形成[dmea‾]xliyhz催化剂时,二甲基氨基乙醇与有机锂的比率可以广泛变化。然而,应该理解,为了形成氢化锂物质,必须相对于摩尔当量的二甲基氨基乙醇使用摩尔过量的有机锂化合物,使得锂-碳键可用于还原成氢化锂物质。可以设想每41摩尔的有机锂试剂采用40摩尔的二甲基氨基乙醇的馈料比;然而,这类馈料比是对昂贵试剂的浪费。优选的馈料比(二甲基氨基乙醇:有机锂)在(1.00:1.05)至约(1:6)范围内,更优选的范围是(1.00:1.10)至约(1:5);甚至更优选的范围是(1.0:1.2)至约(1:4);并且最优选的范围是(1.00:1.40)至约(1:3)。实施例的[dmea‾]4li6h2催化剂由约2摩尔的二甲基氨基乙醇与3摩尔的正丁基锂的馈料比形成。已经发现,用烃溶剂,特别是一定部分的乙苯将约2.0摩尔浓度或更大的有机锂试剂溶液进一步稀释到约≤0.2摩尔浓度有益于(但未必一定)形成更具活性的[dmea‾]xliyhz催化剂。一种解释是,较低浓度的有机锂化合物不太可能存在于有机锂物质的聚合物聚集体中。在本领域中已经很好地确立了烷基锂化合物在苯乙烯和共轭二烯的活跃阴离子聚合反应中的缔合度带来的复杂性(在这方面,参见hsiehh.l.和quirk,r.p.anionicpolymerizationprinciplesandpracticalapplications,marceldekker,1996,newyork,第135至132页,特别是表6.2,第138页)。因为通常供应为相对浓的溶液(≥1.0摩尔浓度的溶液),正丁基锂的缔合度通常为6,其中叔丁基锂和仲丁基锂的缔合度在烃溶剂中通常为4。当形成[dmea‾]xliyhz催化剂时,有机锂化合物用溶剂稀释可导致有机锂试剂的缔合降低,因此导致较低的局部有机锂浓度。尽管我们不希望受理论束缚,但在催化剂形成期间较高的局部有机锂化合物浓度可导致形成活性[dmea‾]xliyhz催化剂的较低活性-即较少可用性的lih-超级聚集体。相对于脂族和脂环族烃的较低pka在抑制有机锂试剂对dmeah的任何不期望的攻击方面也具有一些益处,超过锂dmea-醇盐的最初形成。因此,可以发现,仲丁基锂的使用由于与正丁基锂相比该有机锂化合物的固有的较低缔合状态而比正丁基锂更有效。应该注意的是,据信在氢气氛下形成的[dmea‾]xliyhz催化剂作为聚集体存在,这些聚集体在某些化学计量下可以作为固定分子量的明确定义的物质存在,而其他化学计量或馈料比提供没有明确定义但是作为非均匀聚集体或超聚集体的混合物存在的催化剂。进一步认为,聚叔胺促进剂或任何其他合适的有机路易斯碱(例如,四氢呋喃)的存在可另外稳定某些聚集体或有助于将较大聚集体的其他不太均匀的混合物分解成较小的更具活性的聚集体。因此,催化剂体系的活性可以基于馈料比、催化剂组分和制备催化剂所遵循的方案而在很大程度上变化。用于形成[dmea‾]xliyhz催化剂的分子氢的分压维持在约0.1巴至约300巴或约0.5巴至约12.0巴,或约1.0巴至约10.0巴,或约1.1巴至约5.0巴之间的压力下。可以采用低或高的氢分压,只要为h2从气相到凝相的质量转移提供充分的混合,因此混合对于在相当短的时间内形成[dmea‾]xliyhz催化剂是关键的。形成[dmea‾]xliyhz催化剂所采用的温度维持在约-96℃至约130℃的范围内,更优选在约20℃至约110℃的范围内,且最优选在约30℃至约90℃的范围内。在形成[dmea‾]xliyhz催化剂时并且在随后的初始加热期间,可以将催化剂组分在恰好高于烃溶剂(或溶剂混合物)的熔融温度或进料或将进料的单体的凝固点的温度下组合并反应。在低温(即,-10℃至15℃)且甚至在冷冻条件(-10℃至-126℃)下组合催化剂组分可具有避免或抑制可导致所用的tmeda促进剂和/或dmeah部分分解的锂化反应的益处。因为氮可以(尽管尚未观察到这种情况的证据)潜在地被本发明的[dmea‾]xliyhz催化剂“固定”-即n2可以由本发明的[dmea‾]xliyhz催化剂还原,所以潜在地期望但可能未必消除或者至少使来自反应器顶部空间和系统的n2最少化。可能用存在的其他气体操作,这些气体通常被认为对于活化的氢化物是惰性,例如稀有气体(he、ne、ar)或相对轻质的脂族或环脂族烃(沸点接近或低于反应温度的烃)。在这些惰性气体之中,优选相对轻质的烃(包括由丁基锂试剂形成的任何c4烃),因为这类烃通常可溶于反应介质中,因此随着顶部空间体积减小而不被h2置换,从而或者在恒定反应器压力下进料单体的过程期间以显著改变量降低h2分压。因此,不太期望随着凝相体积增加而在顶部空间中被压缩的惰性气体。然而,在顶部空间体积固定的连续过程中存在诸如稀有气体的这类低溶解性气体也许可能用于某些益处。难以在恒定压力的低正压下操作商业反应器,因此可能有利的是具有目前的低沸点(石油醚)烃,使得所需的h2分压和因此活性可以维持在更高的总反应器压力下。这类轻质烃甚至可以提供某些回流冷却方式的附加益处。当形成[dmea‾]xliyhz催化剂时,tmeda可任选地在还原或氢化物形成过程期间存在,或者任选地在氢化物形成之后添加。tmeda可以有利于在某些条件下形成或[dmea‾]xliyhz催化剂,或者其可能有益于在其使用期间促进[dmea‾]xliyhz催化剂活性,但是未必形成该催化剂所必需。事实上,有一些证据表明在催化剂形成期间甚至痕量的tmeda的存在都会导致催化剂活性降低。据推测,tmeda络合的有机锂试剂可以经历竞争性还原以形成schleyer的超活性纳米尺寸的lih(schleyer,p.v.r.等人,j.org.chem.1987.52,4299;和schleyer,p.v.r.等人,angewchemint.ed.engl.198625465)。已经发现这类纳米lih作为hmaps方法的催化剂(如下所述)非常无效,因此其共形成会降低活性催化剂的滴度。因此,建议在形成[dmea‾]xliyhz催化剂后添加tmeda并非完全必要。同样,建议通过在使用前漂洗催化剂形成反应器和反应器序列以及从回收溶剂中提取或除去tmeda未从[dmea‾]xliyhz催化剂形成步骤中减少(如果不是消除)消除偶然的tmeda方面进行一些努力。甚至可取但没有必要在反应器中形成[dmea‾]xliyhz催化剂,将催化剂转移到已经馈有tmeda或将馈有tmeda的hmaps聚合反应器中。当采用tmeda时,其以锂与tmeda的如下摩尔比(锂:tmeda)存在:约(∞:1)的极限或更实际(10,000:1)至约(1:8),或优选约(5:1)或约(1:5)或甚至更优选(3:1)至约(1:3)。应当理解,在这方面,(∞:1)或更实际地(10,000:1)的馈料比可以表示由于在反应器或馈料线或罐中从有意馈入tmeda的先前运行留下的量而导致意外存在甚至痕量的tmeda促进剂。此外,总锂与tmeda的馈料比大于(1:8)在范围内,然而这一馈料比提供很少(如果有的话)优点并且代表和tmeda促进剂以及任何试剂的不经济使用和/或从反应或产物混合物中除去和/或回收tmeda促进剂所需的额外努力。可用于形成loxlih[dmea‾]xliyhz催化剂的烃溶剂是在反应条件下具有大于分子氢(h2)的pka的任何烃。这类优选溶剂的非限制性实例是环己烷、甲基环己烷,与或不与乙苯一起使用。可以使用其他烃溶剂,只要它们的使用不影响:1)类盐氢化物催化剂、反应性中间体、瞬时活跃的聚合物链和聚合物链分布产物的溶解度;或2)当使用用于hmship或hmaps方法的催化剂时,溶剂不充当具有足够活性使得烃溶剂以约2重量%或更高的水平并入hmaps产物分布中的有机链转移剂。发明详述本申请的另一个实施方案涉及一种用于氢介导的阴离子链转移聚合的方法,其包括在包含分子氢的气氛下将阴离子可聚合单体如乙烯基芳族和/或苯乙烯类单体进料到含有烃溶剂和烃可溶性类盐氢化物催化剂的反应混合物的反应器容器中。可溶性类盐氢化物催化剂包括单独或组合使用的loxsh催化剂。loxsh催化剂方法的优选实施方案具有动力学链长分布(υ),因此聚合度(dpn)和因此由下列关系确定或以其他方式设定的数均分子量(mn):。因此,mn基本上设定而排除其他动力学术语,因此聚合物基本上是仅由氢和单体形成的阴离子链转移聚合物分布,而没有任何添加的有机链转移剂的任何显著并入-至少小于约2重量%,更优选小于1重量%且更优选小于0.1重量%。因此,当单体是苯乙烯时,这类聚合物的mn(排除乙苯含量)由下式给出:其中苯乙烯的摩尔数是苯乙烯的进料量、消耗的氢的摩尔数且其中生成的乙苯的摩尔数小,优选小于产物组合物的10重量%,更优选小于7重量%,且最优选小于5重量%。因此,本发明还涉及一种用于阴离子链转移聚合的loxsh催化剂方法,其包括在反应器容器中在包含分子氢的气氛下将阴离子可聚合单体(例如,乙烯基芳族单体和/或优选苯乙烯类单体)进料到反应混合物中,其中所述反应混合物由以下各物形成:(i)有机锂化合物和/或有机镁化合物;(ii)任选地,聚叔胺化合物;(iii)选自叔氨基醇化合物、叔氨基醚-醇、醚-醇或其组合的极化络合剂;(iv)任选地,碱金属或金属合金和/或固体类盐氢化物和/或类盐金属酰胺;(v)任选地,具有至少一个c-h共价键的芳烃,其pka在甲苯的pka之上2.75pka单位至甲苯的pka之下-4.30pka单位的范围内;(vi)任选地,乙烯基芳族单体;和(vii)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;(viii)分子氢;并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。上面列出的用于形成loxsh催化剂以及用于上述催化剂和或试剂组合物的组分的相同非限制性实例和量可以用于进行loxsh催化剂类盐氢化物引发的氢介导的聚合过程,并且不需要重复。该烃和芳烃溶剂可以相同或不同。这意味着芳烃可以充当芳烃和溶剂两者。考虑到一部分苯乙烯被还原成乙苯,乙苯是商业氢介导的苯乙烯阴离子聚合中的优选组分,因此乙苯是再循环溶剂的可能组分,除非在从用于形成loxsh催化剂以及进行进行本发明的loxsh方法的烃溶剂中分馏共产物乙苯中采取极大的小心、时间和精力。可以设想仅使用乙苯,并且在这种情况下,对于loxsh催化剂形成过程,组分(v)和(vii)将合并成一种组分(或限制)并且是相同的。还可以设想使用不含乙苯的方法,其使用不含乙苯的再循环和/或新鲜的烃溶剂。阴离子可聚合的烃单体可包括一种或多种乙烯基芳族单体,特别是苯乙烯类单体。优选地,乙烯基芳族单体是苯乙烯类单体,诸如苯乙烯,或烷基化苯乙烯单体,诸如甲基苯乙烯的邻位、间位和对位异构体,对异丙基苯乙烯、2,4-二乙基苯乙烯、邻乙基苯乙烯、3,5-二异丁基苯乙烯、2,6-二甲基苯乙烯、2-乙基-4-甲基苯乙烯及其组合。为了形成不含支化分子结构的线性聚合物微结构,苯乙烯是优选的乙烯基芳族单体。烷基化苯乙烯单体在某些工艺条件下本身将表现为链转移剂并导致一定程度的支化和潜在的交联。二乙烯基芳族单体如二乙烯基苯也可用作共聚单体,然而可发生支化和交联。苯乙烯类单体如α-烷基化苯乙烯(例如,α-甲基苯乙烯)通常在链转移条件下不均聚,但可以用作特别是与共轭二烯的共聚单体。然而,应该注意的是,使用α烷基化苯乙烯将导致在聚合物微结构中形成季碳。因此,对于形成为通过亲电芳族取代反应衍生化的底物的苯乙烯聚合物,应避免使用这类α烷基化苯乙烯。上述loxsh催化剂形成过程中采用的分子氢分压维持在约0.5巴至约19.0巴,或约1.5巴至约12.0巴,或约2.5巴至约10.0巴或约3.0巴至约7.0巴的压力下。在该过程期间,当工艺条件需要常规操作并充分混合以维持氢转移至凝相时,可容许氢分压大于约约10.0巴,持续一定的时间。然而,在这类增加的氢分压下的大量时间通常将会导致单体氢化,其中聚合物分子量显著降低。相反地,在涉及氢化钾形式的loxkh催化剂的方法的常规操作期间,可容许氢气压力低于0.1巴(小于1.5psi)。在低氢分压并因此在凝相中具有低h2活性的条件下,无论是在运行过程期间添加还是形成的有机链转移剂的链转移将更显著地竞争。当采用loxlih或loxmgh2及其组合时,在氢质量传递不良的条件下经短时间进料单体导致产生高分子量聚合物链,从而产生具有高分子量尾的更不对称的产物分布(如通过gpc测量值“mw10%高”(道尔顿)显著增加所反映)。需要指出的是,上述分压只有在维持分子氢向凝相的充分质量传递时才有意义,因此分压反映分子氢的凝相活性-即确立了h2向凝相的稳态质量传递。因此,当由于气相与凝相混合不良而导致向凝相的质量传递减少且因此导致差的质量传递时,可以施加高得多的h2分压。因为氮可以(尽管尚未观察到这种情况的证据)潜在地被本发明的类盐氢化物催化剂“固定”-即n2可以被本发明的类盐氢化物还原,并且因为当在半间歇条件下操作时反应器顶部空间被单体进料减少,所以潜在地期望但可能未必消除或者至少最少化来自反应器顶部空间和系统的n2。可能用存在的其他气体操作,这些气体通常被认为对于活化的氢化物是惰性,例如稀有气体(he、ne、ar)或相对轻质的脂族或环脂族烃(沸点接近或低于反应温度的烃)。在这些惰性气体之中,优选相对轻质的烃,因为这类烃通常可溶于反应介质中,因此不被h2置换,从而在恒定反应器压力下进料单体的过程期间显著改变量时不降低h2分压。因此,不太期望随着凝相体积增加而在顶部空间中被压缩的惰性气体。然而,在顶部空间体积固定的连续过程中存在诸如稀有气体的这类低溶解性气体也许可以用于某些益处。难以在恒定压力的低正压下操作商业反应器,因此可能有利的是具有目前的低沸点(例如,石油醚)烃,使得所需的h2分压和因此活性可以维持在更高的总反应器压力下。这类轻质烃甚至可以提供某些回流冷却方式的附加益处。反应混合物和/或方法的温度维持在约20℃至约130℃的范围内,更优选在约40℃至约110℃的范围内,且最优选在约60℃至约90℃的范围内。单体与初始形成的金属氢化物化合物的总馈料摩尔比(单体:金属氢化物)为约(10:1)至约(1000:1),或约(10:1)至约(600:1),或约(40:1)至约(600:1),或约(150:1)至约(360:1)。而形成的金属氢化物的摩尔量被认为等于在催化剂形成反应条件下与pka<h2的所有质子物质反应后保留的有机金属键-有机锂和/或有机镁碳-金属键的摩尔量,其共轭酸的pka>h2。不考虑由于分解反应导致的金属氢化物量的任何减少并且只是最好避免有助于催化剂失活的条件(例如,温度)以及试剂(例如,易于通过有机锂试剂进行金属化和分解的有机物质,诸如某些醚)。在本发明的工艺技术的间歇或半间歇操作中,单体(例如,苯乙烯)随时间进料到反应介质中,因此在蒸气来自进料单体的最初下降或增量的某一时刻形成的单体与类盐金属氢化物的初始比率在数学上接近极限(1:∞)。因此,在下面提供的每个实施例的开始时证明优选描述范围外的馈入的总单体与初始形成的类盐金属氢化物摩尔比是可使用的范围,即,在(1.00:∞至1.00:0.101)的极限范围内的摩尔比,其为约(1:10,000至约9.9:1.0)单体:形成的类盐金属氢化物。然而,通常继续单体进料直至更高的所需单体与金属氢化物的比率完成。实施限于(1:10,000至约9.9:1.0)的馈料摩尔比在本发明的范围内,但仅代表有机锂化合物和/或有机镁化合物的不经济利用。相反,以初始大于约1000:1的单体与类盐金属氢化物化合物的相对摩尔比进料单体变得不可行;导致链转移减少,生成不期望的分子量分布(mwd)的组合物。由于易于形成催化剂、对微结构具有选择性并且最终由于催化剂活性,loxlih催化方法是更优选的方法。在实施例14和15中,证明使用>600:1的单体:金属氢化物馈料比可以实现15,000%的催化剂效率。这两次运行的组合生成二聚物汽提的聚合物分布,其特征在于mw=730,pdn=1.4,分离收率为75%。对于loxsh方法,聚叔胺(pta)促进剂是任选的。因此,当采用单体pta组合物时,pta促进剂以总碱金属和碱土金属与pta的摩尔比(金属:pta)存在,该比率为约(∞:1)的极限或更实际地(10,000:1)至约(1:8),或优选约(5:1)或约(1:5),或甚至更优选(3:1)至约(1:3)。应当理解,在这方面,(1:∞)或更实际地(10,000:1)的馈料比可以表示由于在反应器或馈料线或罐中从有意馈入pta的先前运行留下的量而导致意外存在甚至痕量的pta促进剂。此外,总金属与pta的馈料比大于(1:8)在范围内,然而这一馈料比提供很少(如果有的话)优点并且代表和pta促进剂以及任何试剂的不经济使用和/或从反应或产物混合物中除去和/或回收pta促进剂所需的额外努力。相对于催化剂量的单体进料速率是关于设定聚合物组合物的多分散度pdn,因此如通过mn、mw、mz、pdn、数均标准偏差(σn)和不对称性(nα3)的值测量的总分子量分布(mwd)的决定动力学因素。因此建议在给定的反应器设计或几何形状中以给定的h2活性(或分压)以某些相对速率进料单体。应该清楚的是,单体与催化剂的非常小的相对进料速率(即,小于约15摩尔单体/小时/摩尔活性li)将生成具有一些二聚物的不期望水平的还原(基本上氢化)的单体。此外,生成的组合物具有高不对称值并且不太理想。另一方面,非常高的相对进料速率通常形成更高的分子量分布,这类组合物可以以其他方式经济地生成,而几乎没有链转移。因为loxsh催化剂的分子式未必是决定性的,由于聚集体的形成而未必完全确定,这些催化剂聚集体的分子量未必已知,因此单体(苯乙烯)相对于催化剂的每小时进料速率以催化剂组合物中形成的活性氢化物的量合适地表示。假定每当量摩尔的活性有机锂烷基和/或活性有机镁烷基形成1当量摩尔的类盐氢化物;其中有机锂化合物提供1当量,且有机镁提供2当量。因此,在本发明的实施中,单体与类盐氢化物化合物的每小时进料速率应在约10摩尔至约500摩尔单体/小时/摩尔馈入反应器中的活性类盐氢化物试剂的范围内,或更优选在约65摩尔至约380摩尔单体/小时/摩尔在反应器中初始形成的类盐氢化物的范围内。同样,使类盐氢化物的当量摩尔数等于活性有机锂烷基的摩尔当量和/或初始馈入的活性有机镁烷基的摩尔当量。同样,活性有机锂烷基和/或活性有机镁烷基的摩尔当量是指在与反应混合物中存在的pka小于h2的任何和所有质子物质反应后留下的有机锂烷基的量和/或镁烷基基团的量。在单体进料过程期间反应混合物的温度维持在约20℃至约130℃范围内,或在约40℃至约99℃范围内,或在约60℃至约90℃范围内。可以想象,在整个运行期间或在该运行的一部分期间可以采用更高的温度;然而,最好避免加速催化剂的任何分解和/或导致从聚合物链中消除氢化物和形成以不饱和键封端的链长的温度。这类氢化物消除终止反应的量应随温度和催化剂组成而变化。在形成loxsh催化剂时且在初始加热期间,催化剂可以在刚好高于烃溶剂(或溶剂混合物)的熔融温度或正进料的单体的凝固点的温度下组合。在低温(即,-10℃至15℃)和甚至在冷冻条件(-10℃至-126℃)下组合催化剂组分可以具有避免或抑制锂化或其他可导致所用的聚叔胺促进剂和/或极化络合剂的部分分解的金属化反应的益处。然而,可能最好避免导致类盐氢化物催化剂或其前体络合物和试剂沉淀的条件。单体在反应介质中的所需分散水平将取决于在整个运行过程中氢气从气相和/或氢气进料输送到凝相的效率。理想地,可以设计并配置商业规模、中试规模和甚至实验室规模的反应器,使得在整个单体进料过程中氢气从气相转移到凝相基本上是均匀的。在相之间的这类均匀氢输送下,期望通过进料单体使单体还原为其饱和类似物最小化,使得反应器中存在局部高浓度。在实验室或小型中试规模的反应器中,通过采用非常高的相对单体与催化剂进料速率和比率以及使用相对低的进料速度来实现这种局部高单体浓度。在大型商业设备中,将单体进料到反应区,该反应区可以与反应混合物的主体物理分开或分离(即,循环泵回路)。尽管loxlih催化剂由2-[2-(二甲基氨基)乙氧基]乙醇(dmaeoe)形成,但本发明的这些hmship方法的类盐氢化物催化剂的优点在于这类催化剂在工艺条件下似乎相当稳定并且不降低催化剂活性或生成衍生自所用的聚叔胺促进剂或氨基醇盐极化络合剂的杂质。在这些条件下,还原单体的产量保持远低于馈入单体总量的10%。二聚物含量也可以保持低于产物分布的12%,因此基于馈入的单体,三聚物及更高级的多聚物的收率可以远远超过80%至90%。完成loxsh催化剂工艺单体进料和反应后,如所指示,例如通过在恒定热通量下的工艺温度的快速降低和/或h2吸收的终止,将反应混合物维持在氢气压力下,然后转移到用于猝灭和水洗的洗涤反应器中。向洗涤反应器中馈入水(有或没有无机酸如h2so4和/或有机酸如乙酸)。另外,可向洗涤反应器预先馈入任选另外量的溶剂,优选烃溶剂。猝灭可以在冷却下或在环境温度至高达在洗涤反应器的压力条件下烃溶剂与水形成共沸物的温度下进行。将产物水洗以除去碱金属盐和至少一部分的pta促进剂(如果存在的话)和极化络合剂。在非常酸性的条件下,用由酸形成的碱金属和碱土金属盐几乎完全除去这类试剂。在使用一当量或更少的酸的碱性条件下,pta促进剂(如果存在的话)和极化络合剂在有机反应混合物和水洗液之间分配。继续水洗直至获得所需ph的排出洗涤水。在碱性条件下,ph9至ph11表明已经除去所有的碱金属和碱土金属盐。在酸性条件下,ph6至ph8(取决于洗涤水的碱度)表明所有酸性物质都已经被除去或至少被中和。当洗涤液被认为是完全溶剂且任何剩余的聚叔胺促进剂的一部分以及任何剩余的极化络合剂(如果存在的话)和单体还原产物优选从反应混合物中分离并回收,由此最后痕量的水也从反应混合物中共沸除去。应继续该分离操作直至所得产物混合物的单体还原产物含量小于约0.1重量%。一些应用期望通过降低单体二聚物含量进一步改进并成型产物分布。对于高沸点的二聚物,这使用刮膜蒸发器容易地进行。本发明涉及一种进行氢介导的苯乙烯阴离子聚合(hmaps)的方法,该方法在某些优选条件下以共收率形成新型且有益的低分子量阴离子链转移聚合物分布,其不对称性低,具有非常纯的“头对尾”微结构。该方法的特征在于在包含分子氢的气氛下将苯乙烯单体进料到含有[dmea‾]xliyhz催化剂的合适溶剂中,其中来自分子氢的链转移是决定包含乙苯共产物的所得产物分布的数均分子量(mn)的机理的重要分量。因此,hmaps产物分布的数均分子量由下式给出:其中[苯乙烯]是苯乙烯的进料总量,并且[h2]是在一段时间内消耗的氢的总量,无论该时间是瞬时的还是整个聚合反应的时间。当单体时,由这一方法形成的产物分布在下文中指定为hmaps分布。设定以hmapsm分子量分布而言的形状(即,mn,mw,mz;pdn、σn和不对称性),并由此通过在特定催化剂浓度和氢分压或活性下苯乙烯单体与催化剂的相对进料速率控制。本发明还涉及一种用于阴离子链转移聚合的方法,其包括在具有式[dmea‾]xliyhz的氢介导的链转移聚合催化剂的反应器容器中在包含分子氢的气氛下将乙烯基芳族单体和/或优选苯乙烯类单体进料到反应混合物中,其中所述催化剂由以下物质的接触过程形成:(i)约y当量的有机锂化合物;(ii)任选地,tmeda化合物;(iii)约x当量的二甲基氨基乙醇;(iv)任选地,乙苯;(v)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;和(vi)分子氢,其中形成的氢化物的量z由公式z=y-x给出,并且x、y和z是大于零的正实整数或分数;其中所述式可以进一步包含n,n,n’,n’-四甲基乙二胺(tmeda)配体络合物,即[dmea‾]xliyhz•xtmeda,摩尔比为x摩尔tmeda/摩尔催化剂[dmea‾]xliyhz,其中x=0.0001至约8.0。因此,本发明还涉及一种用于阴离子链转移聚合的hmaps方法,其包括在反应器容器中在包含h2的气氛下将苯乙烯单体进料到反应混合物中,其中所述反应混合物含有具有化学式[dmea‾]xliyhz的催化剂,其中所述催化剂由使以下物质接触的过程形成:(i)约y当量的有机锂化合物和/或有机镁化合物;(ii)任选地,tmeda化合物;(iii)约x当量的二甲基氨基乙醇;(iv)任选地,乙苯;(v)pka大于h2的烃溶剂;其中芳烃和烃溶剂可以相同或不同;和(vi)分子氢;其中形成的氢化物的量z由公式z=y-x给出,并且x、y和z是大于零的正实整数或分数并且其中包含所述催化剂或试剂的氢化物的溶解度为至少约0.0080摩尔/升。上面列出的用于形成[dmea‾]xliyhz催化剂以及用于上述催化剂和或试剂组合物的组分的相同非限制性实例和量可以用于进行[dmea‾]xliyhzhmaps过程,并且不需要重复。可使用的烃溶剂是在反应条件下具有大于分子氢(h2)的pka的任何烃。优选的这类溶剂的非限制性实例是环己烷、甲基环己烷,与或不与乙苯一起使用。可以使用其他烃溶剂,只要它们的使用不影响:1)类盐氢化物催化剂、反应性中间体、瞬时活跃的聚合物链和聚合物链分布产物的溶解度;或2)充当具有足够活性使得烃溶剂以约2重量%或更高的水平并入hmaps产物分布中的有机链转移剂。在进行本发明的hmaps方法以形成本发明的最期望的hmaps分布时,相对于氢化锂的单体进料通常在约(10:1)至约(1000:1)的范围内,优选在约(50:1)至约(800:1)范围内,且最优选在(100:1)至约(600:1)范围内,在运行开始时氢化锂浓度为约200ppm至约750ppm,并且在该方法的半间歇操作的运行结束时,氢化锂浓度为65ppm至约350ppm,这取决于苯乙烯单体的进料总量。对于连续操作模式,优选操作该方法使得在整个操作过程中氢化锂浓度在约200ppm和约500ppm之间。在本发明的工艺技术的间歇或半间歇操作中,将单体(例如,苯乙烯)随时间进料到反应介质中,因此在蒸气来自进料单体的最初下降或增量的某一时刻形成的单体与[dmea‾]xliyhz催化剂的初始比率在数学上接近极限(1:∞)。因此,在下面提供的每个实施例的开始时证明优选描述范围外的馈入的总单体与初始形成的类盐氢化锂摩尔比是可使用的范围,即,在(1.00:∞至1.00:0.101)的极限范围内的摩尔比,其为约(1:10,000至约9.9:1.0)单体:形成的[dmea‾]xliyhz催化剂。然而,通常继续单体进料直至更高的所需单体与氢化锂的比率完成。实施限于(1:10,000至约9.9:1.0)的馈料摩尔比在本发明的范围内,但仅代表用于形成[dmea‾]xliyhz催化剂的有机锂化合物的不经济利用。相反,以初始大于约1000:1的单体与[dmea‾]xliyhz催化剂的相对摩尔比进料单体由于粘度水平而变得不可行;导致链转移减少,由此生成不期望的分子量分布(mwd)的组合物。[dmea‾]xliyhz催化的工艺技术的特征在于易于形成催化剂,在微结构方面具有选择性,并且最终具有高催化剂活性。在实施例14和15中,证明使用>600:1的单体:氢化锂馈料比可以实现15,000%的催化剂效率。这两次运行的组合生成二聚物汽提的聚合物分布,其特征在于mw=730,pdn=1.4,分离收率为75%。相对于[dmea‾]xliyhz催化剂量的单体进料速率是关于设定聚合物组合物的多分散度pdn且因此如通过mn、mw、mz、pdn、数均标准偏差(σn)和不对称性(nα3)的值测量的总分子量分布(mwd)的决定动力学因素。因此建议在给定的反应器设计或几何形状中以给定的h2活性(或分压)以某些相对速率进料单体。应该清楚的是,单体与催化剂的非常小的相对进料速率(即,小于约15摩尔单体/小时/摩尔活性li)将生成具有一些二聚物的不期望水平的还原(基本上氢化)的单体。此外,生成的组合物具有高不对称值并且不太理想。另一方面,较高的相对进料速率通常形成较高的分子量分布,其中乙苯和苯乙烯二聚物的收率较低。在汽提乙苯(收率低至4%至6%)和二聚物(收率低至8%至12%)后可以容易地制备mw在850至1050范围内的hmaps组合物,收率为约82%至约90%。在单体进料过程期间反应混合物的温度维持在约20℃至约130℃范围内,或在约40℃至约99℃范围内,或在约60℃至约90℃范围内。可以想象,在整个运行期间或在该运行的一部分期间可以采用更高的温度;然而,最好避免加速催化剂的任何分解和/或导致从聚合物链中消除氢化物和形成显著水平的用不饱和键封端的链长分布的温度。这类氢化物消除终止反应的量可以随温度和催化剂组成而变化。单体在反应介质中的所需分散水平将取决于在整个运行过程中氢气从气相和/或氢气进料输送到凝相的效率。理想地,可以设计并配置商业规模、中试规模和甚至实验室规模的反应器,使得在整个单体进料过程中氢气从气相转移到凝相基本上是均匀的。在相之间的这类均匀氢输送下,期望通过进料单体使单体还原为其饱和类似物最小化,使得反应器中存在局部高浓度。在实验室或小型中试规模反应器中,通过采用非常高的相对单体与催化剂进料速率和比率以及使用由进料速率(体积/秒)和进料单体的面积(由圆柱形进料尖端的半径设定)(即,cc/sec除以平方厘米)控制的相对低的进料速度实现这类局部高单体浓度。在大型商业设备中,可以将单体进料到反应区,该反应区可以与反应混合物的主体物理分开或分离(即,循环泵回路)。[dmea‾]xliyhz催化剂的优点在于,这类催化剂在工艺条件下似乎相当稳定,并且不会遇到催化剂活性降级的问题,因此不会导致形成衍生自所用的聚三胺促进剂或氨基醇盐极化络合剂的杂质。在hmaps工艺条件下,还原乙苯的形成保持远低于馈入单体总量的10%。二聚物含量也可以保持低于产物的12%。基于馈入的总单体,汽提到小于2%,优选小于1%的苯乙烯二聚物含量且因此包含>98%三聚物及更高级多聚物的hmaps分布的收率可以远远超过80%至90%。一旦组分以所需的馈料比组合,催化剂并且如果需要进一步活化,催化剂就准备用于本发明的氢介导的阴离子聚合方法。因此,苯乙烯单体在约0.001巴至约10.0巴,或约0.3巴至约6.8巴,或约0.5巴至约5.2巴,或约1.0巴至约4.2巴的氢分压下进料到[dmea‾]xliyhz催化剂中。在该过程期间,当工艺条件需要常规操作并充分混合以维持氢转移至凝相时,可容许氢分压大于约10.0巴,持续一定的时间。然而,在这类增加的氢分压下的大量时间通常将会导致单体氢化,其中聚合物分子量显著降低,乙苯收率增加。相反,在该过程的常规操作期间可容许氢气压力低于约0.1巴(小于1.5psi),但是将导致高不对称性的组合物具有分子量非常高的尾部,该尾部具有高于如通过gpc测量的约2000道尔顿的10%高分子量。当反应条件导致氢向凝相的质量转移差时,也发生具有高不对称性的这类组合物的形成。当在产生粘度增加的反应介质的不充分混合条件下短时间进料单体时,观察到以产物分子量分布为特征的这类高分子量尾部的形成。增加的粘度使得氢的质量传递越来越低效,导致粘度甚至更大程度地增加。可导致粘度增加的反应条件有:1)反应温度;和/或2)低于最佳催化剂浓度;和/或3)低于最佳单体与催化剂的馈料比;和/或4)过高的局部单体浓度:和/或5)由于反应器几何形状/设计差而导致混合变得低效时的进料时间。因此,需要指出的是,上述分压只有在维持分子氢向凝相的充分质量传递时才有意义,因此分压反映分子氢的凝相活性-即,确立了h2向凝相的稳态质量传递。因此,当由于气相与凝相混合不良而导致向凝相的质量传递减少且因此导致差的质量传递时,可以施加高得多的h2分压。然而,如果粘度变得太大,则增加的混合通常导致泡沫的形成,向凝相的甚至更低效的质量传递。完成[dmea‾]xliyhz催化剂hmaps工艺苯乙烯单体进料和反应后,如所指示,例如通过在恒定热通量下的工艺温度的快速降低和/或h2吸收的终止,将反应混合物维持在氢气压力下,然后转移到用于猝灭和水洗的洗涤反应器中。向洗涤反应器中馈入水(有或没有无机酸如h2so4和/或有机酸如乙酸)。另外,可向洗涤反应器预先馈入任选另外量的溶剂,优选烃溶剂。猝灭可以在冷却下或在环境温度至高达在洗涤反应器的压力条件下烃溶剂与水形成共沸物的温度下进行。将产物水洗以除去碱金属锂盐和至少一部分的tmeda促进剂(如果存在的话)和极化络合剂。在非常酸性的条件下,用由酸形成的碱金属和碱土金属锂盐几乎完全除去这类试剂。在使用一当量或更少的酸的碱性条件下,tmeda促进剂(如果存在的话)和dmeah试剂在有机反应混合物和水洗液之间分配。继续水洗直至获得所需ph的排出洗涤水。在碱性条件下,ph9至ph11表明已经除去所有的碱金属和碱土金属锂盐。在酸性条件下,ph6至ph8(取决于洗涤水的碱度)表明所有酸性物质都已经被除去或至少被中和。有时可能期望在酸性洗涤条件下添加少量(20mg,两升运行)的表面活性剂如十二烷基硫酸钠以破坏任何乳液或胶束形成当洗涤液被认为是完全溶剂且任何剩余的聚叔胺促进剂的一部分以及任何剩余的极化络合剂(如果存在的话)和乙苯共产物优选从反应混合物中分离并回收,由此最后痕量的水也从反应混合物中共沸除去。应继续该分离操作直至所得产物混合物的乙苯共产物含量小于约0.1重量%。通过优选用刮膜式蒸发器蒸馏降低苯乙烯二聚物含量来进一步成型hmaps分布,实现基本上完全去除乙苯的改性。与其他现有技术(例如,epo741147)相比,以原位形成锂醇盐能力为特征的hmaps方法既是实验室方便的,又是形成烃可溶性类盐金属氢化物催化剂的主要商业优势。因此,当形成loxlih和loxmgh2催化剂时,原位形成锂和/或镁醇盐试剂前体:(1)避免处理易燃空气和水分反应性固体;(2)消除金属醇盐溶解在大摩尔过量的聚叔胺促进剂的浓缩物中;(3)在溶剂和其他试剂的再循环之前,消除从工艺流中除去痕量的副产物醇的需要,以及(4)大大降低并且在一些实施方案中消除聚叔胺促进剂的需要量。loxlih和loxmgh2催化剂似乎都更均匀地溶于催化剂形成和/或聚合反应混合物中。因此,认为运行之间的可变性得以显著改善-更可重现-因此产生比实施例38和39的sash方法或实施例40的hash方法更稳健的商业方法。loxlih和loxmgh2催化剂似乎也在聚合反应器和其他相关设备的壁和内部部件上留下很少量的不期望的固体。反应器表面上的不溶性催化剂沉积是与现有技术有机链转移方法相关的显著复杂性,其中催化剂是在诸如氮气的惰性气氛下由诸如正丁基锂、叔丁醇钾、tmeda和乙苯的试剂形成。loxlih和loxmgh2催化剂以及它们催化的氢介导的阴离子链转移方法的意外优势是这些催化剂和方法提供具有微结构的纯链转移聚苯乙烯组合物,其没有断裂聚合和链异构化杂质或杂质分布。这已经通过由气相色谱分析前4至6种低聚物通过实验证明(参见图3和图4,其比较loxlihps低聚物微结构与现有技术的乙苯链转移聚合ps衍生的低聚物的微结构)。loxkh催化剂方法以及hash和sash方法通常生成具有不太期望的微结构的聚苯乙烯组合物(参见,图5至图7),这也是现有技术所共有的(也参见图12至图14)。因此,loxlihps和loxmgh2ps组合物在形成衍生自另外的化学如对产物分布进行的芳族亲电取代反应的商业产品方面非常有利。应当理解,明智地选择催化剂的氨基醇组分-包括光学活性的氨基醇-以及进一步的实验,本发明的实践者可以发现控制其他微结构特征如乙烯基芳族聚合物的立构规整度的方法。因此,本发明的另一个实施方案是用类盐氢化物引发并且具有聚合物微结构的阴离子链转移苯乙烯类聚合物分布,如由上面的聚合物聚苯乙烯结构12所示(具体地对于但不限于苯乙烯示出),该聚合物微结构是大于97%的头对尾微结构,更优选大于98%的头对尾微结构,且最优选大于99%的头对尾微结构。换句话说,本发明的阴离子链转移苯乙烯类聚合物组合物是通过将类盐氢化物加成到苯乙烯类单体上引发,并且具有如下链长分布,其中在该聚合物微结构中小于3.0%,更优选小于2.0%且最优选小于0.8%的聚合物链具有一个或多个季碳。另外,上面的氢化物引发的聚(苯乙烯类)结构(再次具体地对于但不限于苯乙烯示出)12的所述分布包含小于3%,更优选小于2%,甚至更优选小于1%,且最优选小于0.2%至低于链长分布的检测极限的量的由断裂聚合过程产生的共产物分布,其中链长分布的微结构和纯度下面由自所述氢介导的类盐氢化物引发的苯乙烯或聚(苯乙烯类)或聚(乙烯基芳族)分布获得的最低分子量链的气相色谱分析确定,其中上述聚合物结构的n=0至n=4:最优选的初始形成的氢介导的类盐氢化物引发的苯乙烯分布仅由苯乙烯单体和氢形成,并具有上述结构(12)的链长分布。所述链长分布由相对摩尔含量的统计数均分布中的i-1个离散聚合物链长组成,其中i是并入给定离散聚合物链中的单体总数。数i是从i=2到i=i的正整数。因此,对于(链-1),当n=0时(苯乙烯二聚物),i=2;对于(链-2),当n=1时(苯乙烯三聚物),i=3;对于(链-3),n=2(苯乙烯四聚物),i=4;对于(链-4),当n=3时(苯乙烯五聚物),i=5;对于(链-5),当n=4时(苯乙烯六聚物),i=6;......并且对于(链-(i-1)),当n=i-2时,i=i。因此,第(i-1)个离散聚合物链是最长链长的离散聚合物链。我们已经发现,通常,本发明的聚合物组合物的gpcmwd分析结果可以用gamma概率密度函数(pdf)合理地建模。然而,更重要的是,我们已经发现由除基于单金属锂的loxlih催化剂以外的催化剂形成的组合物通常用betapdf更精确地建模。最重要的是,loxlihpsgpcmwd结果由weibullpdf精确地建模,这表明,对于loxlih催化体系,分子量分布仅由链转移设定,而没有显著的无活性聚合物链再生,并且可以表明没有活化,乙苯作为有机链转移剂参与或并入形成聚合物分布中。当苯乙烯是单体时,本发明的链长分布的分子量分布的特征在于,mn在315道尔顿至934道尔顿的范围内;mw在约392道尔顿至约1705道尔顿的范围内;并且mz在约512道尔顿至2930道尔顿的范围内;pdn在1.24至1.82的范围内;标准偏差在156道尔顿至849道尔顿的范围内,并且不对称性在1.40至约3.00的范围内。更优选的组合物具有如下分子量分布,其中mn在410道尔顿至680道尔顿的范围内;mw在约553道尔顿至约1205道尔顿的范围内;并且mz在约745道尔顿至1950道尔顿的范围内;pdn在1.29至1.82的范围内;标准偏差在257道尔顿至600道尔顿的范围内,并且不对称性在1.50至约2.60的范围内。最优选的组合物具有如下分子量分布,其中mn在444道尔顿至683道尔顿的范围内;mw在约600道尔顿至约1150道尔顿的范围内;并且mz在约798道尔顿至1768道尔顿的范围内;pdn在1.35至1.68的范围内;标准偏差在263道尔顿至565道尔顿的范围内,并且不对称性在1.50至约2.31的范围内。本发明的优选的非共混组合物基本上仅(如果不是唯一的话)由苯乙烯组成,具有大于97重量%的“头对尾”微结构,并且其链长分布通过除去一部分的最低分子量链进一步成形或改性。除去较低分子量的链,特别是苯乙烯二聚物-如从所有其他算术平均值(例如,平均积分点)中除去最低值或最低值的一部分-产生新的平均值,其中总分子量分布增加。因此,本发明的优选的改性分子量分布将与未改变的分布重叠,但可能不在上面指定的分子量分布或分子量参数的范围内,因为简单的数值结果已通过除去分布的较低分子量分数的一部分而改变。因此,二聚物含量已经降低但仍然存在并且占整个分布的约0.1重量%至约1.0重量%(如通过gpc分析确定)的优选组合物具有如下分子量或链长分布,其中mn在407道尔顿至1018道尔顿的范围内;mw在约487道尔顿至约1741道尔顿的范围内;并且mz在约579道尔顿至2938道尔顿的范围内;pdn在1.40至1.71的范围内;标准偏差在180道尔顿至858道尔顿的范围内,并且不对称性在1.31至约3.016的范围内。更优选的组合物具有如下分子量分布,其中mn在494道尔顿至788道尔顿的范围内;mw在约623道尔顿至约1278道尔顿的范围内;并且mz在约782道尔顿至1964道尔顿的范围内;pdn在1.26至1.62的范围内;标准偏差在253道尔顿至621道尔顿的范围内,并且不对称性在1.40至约2.40的范围内。最优选的组合物具有如下分子量分布,其中mn在521道尔顿至737道尔顿的范围内;mw在约661道尔顿至约1202道尔顿的范围内;并且mz在约827道尔顿至1783道尔顿的范围内;pdn在1.27至1.63的范围内;标准偏差在270道尔顿至586道尔顿的范围内,并且不对称性在1.40至约2.50的范围内。需要指出的是,将统计分布组合在一起的共混操作可以产生非统计分布,其中为pdn、标准偏差提供的约束将不适用。然而,这些共混物在本发明的范围内,因为它们是通过组合本发明的组合物形成并且由本发明形成的。因此,本发明的另一个实施方案是指定为hmaps分布的loxlihps分布。对于给定的催化剂组成、催化剂浓度和氢气压力,这类hmaps分布具有mn≈[苯乙烯]/[h2]和mw和mz的设定值或者苯乙烯与催化剂的相对进料速率。hmaps分布由氢化锂引发,由来自氢的质子终止并且具有如下聚合物微结构,如由上面的聚合物聚苯乙烯结构12所示,其为大于97%的头对尾微结构,更优选大于98%的头对尾微结构,并且最优选大于99%的头对尾微结构。换句话说,本发明的hmaps聚合物组合物是通过将氢化锂加成到苯乙烯单体上引发,并且具有如下链长分布,其中在该聚合物微结构中小于3.0%,更优选小于2.0%且最优选小于0.8%的聚合物链具有一个或多个季碳。另外,上面12的hmaps分布包含小于3%,更优选小于2%,甚至更优选小于1%且最优选小于0.2%至低于链长分布的检测极限的量的由断裂聚合过程产生的共产物分布,其中该链长分布的微结构和纯度下面由自hmaps分布获得的最低分子量链的气相色谱分析确定,其中上面聚合物结构12的n=0至n=2。最优选的初始形成的hmaps分布具有以下链长分布:由相对摩尔含量的统计数均分布中的i-1个离散聚合物链长组成,其中i是并入给定离散聚合物链中的单体总数。数i是从i=2到i=i的正整数。因此,对于(链-1),当n=0时(苯乙烯二聚物),i=2;对于(链-2),当n=1时(苯乙烯三聚物),i=3;对于(链-3),n=2(苯乙烯四聚物),i=4;对于(链-4),当n=3时(苯乙烯五聚物),i=5;对于(链-5),当n=4时(苯乙烯六聚物),i=6;......并且对于(链-(i-1)),当n=i-2时,i=i。因此,第(i-1)个离散聚合物链是最长链长的离散聚合物链。我们已经发现,通常,本发明的聚合物组合物的gpcmwd分析结果可以用gamma概率密度函数(pdf)合理地建模。然而,更重要的是,我们已经发现hmaps分布通常由weibullpdf非常好地建模。这随后被解释为意味着聚合过程限于以下步骤:i)引发;ii)链增长;和iii)链终止几乎排他地(如果不是排他地的话)由氢介导实现,即乙苯没有作为链转移剂的动力学活性。本发明的hmaps组合物的分子量分布通过gpc(uv检测器)分析表征,其中mn在400道尔顿至800道尔顿的范围内;mw在约600道尔顿至约1200道尔顿的范围内;pdn在约1.35至约1.75的范围内;标准偏差在约270道尔顿至约550道尔顿的范围内,并且mw10%高小于约3300道尔顿。更优选的组合物为hmaps聚合物,如通过gpc(uv检测器)分析所测量表征其分布,其中mn在约400道尔顿至约800道尔顿的范围内;mw在约600道尔顿至约1200道尔顿的范围内;mz在约750道尔顿至1500道尔顿的范围内;pdn在约1.35至约1.75的范围内;标准偏差在约270道尔顿至约550道尔顿的范围内,不对称性在约1.60至约2.2的范围内;并且mw10%高小于约2400道尔顿。本发明的优选的hmaps分布基本上仅(如果不是唯一的话)由苯乙烯组成,具有大于97重量%的“头对尾”微结构,并且其链长分布通过除去一部分的最低分子量链进一步成形或改性。除去较低分子量的链,尤其是苯乙烯二聚物,对于分子量分布的每个时刻产生新的更高值(即mn、mw和mz),因此导致总mwd增加和改变。因此,本发明的优选的改性分子量分布将与未改变的分布重叠,但可能不在上面指定的分子量分布或分子量参数的范围内,因为简单的数值结果已通过除去分布的较低分子量分数的一部分而改变。因此,二聚物含量已经降低但仍然存在并且占整个分布的约0.1重量%至约1.5重量%(如通过gpc分析uv检测器确定)的优选hmaps分布具有如下分子量或链长分布,其中mn在约500道尔顿至约800道尔顿的范围内;mw在约650道尔顿至约1200道尔顿的范围内;并且mz在约900道尔顿至约1500道尔顿的范围内;pdn在约1.25至约1.70的范围内;标准偏差在约280道尔顿至约600道尔顿的范围内;不对称性在约1.45至约3.20的范围内;并且mw10%高在约1500道尔顿至约3500道尔顿的范围内。需要指出的是,将统计分布组合在一起的共混操作可以产生非统计分布,其中为pdn、标准偏差提供的约束将不适用。然而,这些共混物在本发明的范围内,因为它们是通过组合本发明的组合物形成并且由本发明形成的。因此,本发明还涉及由纯聚苯乙烯组合物的亲电芳族溴化形成的聚合物阻燃剂组合物。所述溴化聚苯乙烯组合物通过需要在溶剂中用溴和溴化催化剂进行溴化的方法或者由用类盐氢化物引发的苯乙烯单体的阴离子聚合形成的聚苯乙烯组合物的其他已知溴化方法制备。因此,本发明提供包含上述hmaps分布的溴化聚苯乙烯的阻燃剂组合物,其中该组合物:(i)溴含量在约73重量%至约77重量%范围内;(ii)低于50ppm的检测限且不高于约1000ppm的在300℃下的热hbr值,该重量%和ppm值基于组合物的总重量;在大于约355℃至约375℃的温度下发生的热重(tga)重量损失为5%;和玻璃化转变温度在约110℃至约155℃的范围内。在本说明书或其权利要求书任何地方的化学名称或化学式所指代的组分无论以单数提及还是以复数提及,都鉴别为其呈在与化学名称或化学类型指代的另一物质(例如另一组分、溶剂等)接触之前所呈现的形式。它关注的不是在所得混合物或溶液中发生了(如果有)什么化学变化、转化和/或反应,因为这些变化、转化和/或反应是将规定的组分在本案所要求的条件下集合在一起的自然结果。因此,组分被鉴定为结合执行所需操作或形成所需组合物聚集在一起的成分。而且,尽管下文权利要求书可以现在时(“包括(comprises)”、“是(is)”等)提及物质、组分和/或成分,但该提及是指物质、组分或成分呈它在根据本发明即将与一种或多种其他物质、组分和/或成分第一次接触、共混或混合之前时所呈现的形式。因此,物质、组分或成分在如果根据本案和化学家的一般技术进行的接触、掺合或混合操作期间可能通过化学反应或转化丢失它原始的本性的事实并无实际意义。本文描述并要求保护的发明不限于本文公开的具体实施例和实施方案的范围,因为这些实施例和实施方案旨在说明本发明的几个方面。任何等同的实施方案旨在属于本发明的范围之内。实际上,除了本文所显示并描述的那些之外,本发明的各种修改对于本领域技术人员来说将从前面的描述中变得显而易见。这些修改也意图属于随附权利要求书的范围内。以下实施例说明了本发明。然而,应该理解,如本文中充分描述的并如权利要求中所述的本发明旨在不受以下实施例的细节限制。实施例使用的通用设备用于hmship过程的装置如下。具有热电偶、底部排液阀、冷却盘管、热油夹套和两个或三个或四个节距叶片涡轮叶轮(如下面所述并在实施例中说明布置每个叶轮)的316不锈钢2升帕尔高压釜进一步配备了活塞泵、隔膜泵、氮气吹扫的250毫升不锈钢装料容器、良好校准的高压计量泵和具有0.02”或0.01”或0.007”内径末端部分的1/16英寸外径表面下单体进料管线(如所述或如实施例中另有说明)。搅拌器上的磁性驱动器连接到高速气动马达并且通常操作(除非在实施例中另有说明),使得搅拌器叶轮在聚合期间以1130±25rpm的速率旋转。将高压釜放空到油封鼓泡器和/或具有底部排放口的6升带油夹套的褶皱洗涤容器并且配备成适于顶部搅拌和蒸馏。高压釜的底部排液阀和浸入管取样口都连接到洗涤容器,以直接转移未猝灭的反应混合物。将散装溶剂(例如,环己烷或甲基环己烷或乙苯或自先前运行中回收的它们的混合物)通过活塞泵经馈料容器馈入反应器中。催化剂组分(例如,tmeda/叔丁醇钾/溶剂溶液和丁基锂)通过装料容器在用精细计量的vernier手柄针阀控制的流速下单独地馈入反应器中。将馈料容器的内容物在最小氮背压下压力转移到具有氮气或氢气或氢气/氮气气氛的高压釜中。苯乙烯通过高压计量泵经碱性氧化铝柱(1或2个0.5”外径柱,每个具有11.0g60-325目al2o3)进料,从而以预定的恒定速率除去抑制剂。将氢气进料到顶部空间和/或表面下并维持在期望的压力下。高压釜反应器用温度设定点为或略高于(+1℃至+3℃)期望的反应温度的油加热,并且一旦反应器控制器按装置的制度操作(当在环境温度下开始时,通常在单体进料的前20至30分钟后),反应温度就紧紧维持在预定的设定点下。因此,反应温度可能具有短暂的温度偏移,这通常不会高于期望的设定点温度5℃以上。在本发明的开发过程期间,利用4种包括两、三或四节距叶片涡轮叶轮的单独构造(下面的构造i-iv)或布置:i.两节距叶片,距反应器顶部,第一个6.25”且第二个10.0”;ii.三节距叶片,距反应器顶部,第一个6.0”、第二个8.0”且第三个10”;iii.三节距叶片,距反应器顶部,第一个5.0”、第二个7.0”且第三个10”;iv.四节距叶片,距反应器顶部,第一个4.0”、第二个6.0”,第三个8”且第四个10”。2升高压釜是深度为10英寸的圆筒,因此每英寸表示200毫升的体积。具有3个叶轮的构造ii和iii导致在整个进料过程中均匀吸收氢,只要进料受到限制使得反应器中的总体积不会显著高于顶部叶轮在质量传递中变得无效的料位。使用4节距叶片叶轮的构造iv是用于在帕尔2升反应器中操作本发明的优选构造-特别是用本发明的loxsh催化剂,该构造允许充分使用反应器的体积,蒸气空间向凝相均匀质量传递,因此在整个苯乙烯单体进料中吸收氢气。由于聚合反应器的体积为2000ml,最大工作体积为1750ml,并且初始形成的反应混合物的体积通常为400ml至600ml,所述可安全进料的苯乙烯的最大体积在1350ml至1150ml的范围内(不考虑聚合时的温度或密度变化)。因此,进料这样体积的苯乙烯(1150ml至1350ml)被认为是该反应器构造的苯乙烯或单体的完全馈料。术语苯乙烯的完全馈料或苯乙烯的部分馈料或者用以暗示苯乙烯的馈料分数部分的任何术语或其他短语是对如上所述的设备的限制或约束,而绝不代表对在具有不同反应器几何形状或构造或操作模式(间歇、半间歇、半连续、连续、回混或堵塞流动是在本发明范围内的所有模式和/或构造)的反应器系统中本发明的方法和实施的限制。下面列举的实施例代表本发明的间歇或半间歇操作。显然,本领域普通技术人员可以采用这些实施例的教导并将本发明的应用扩展到包括需要连续操作而有和/或没有某种程度的反混的操作模式,因此这类模式完全在本发明的范围内。当在氮气氛下对聚合反应器进行馈料时,通过加压将高压釜反应器吹扫至少3次,然后用65psigh2放空(65psig放空至0psig)。然后将聚合反应器加压至期望的h2压力。如果对含有氢气氛的反应器进行反应器馈料,则通常将反应器加压并用50psigh2放空2次。苯乙烯(99%,acros)、tmeda(aldrich)、2-甲氧基甲醇(99.9%aldrichhplc级)、2-n,n-二甲基乙醇胺(99.5%,aldrich)、2-[2-(二甲基氨基)乙氧基]乙醇(98%,aldrich)、氢化钾(30%在矿物油中,aldrich)、庚烷中的1.0m二正丁基镁和正丁基锂(在环己烷中2m(aldrich)各自按供应商的原样使用。将无水环己烷甲基环己烷和乙苯(全部来自aldrich)在惰性干燥氮气氛下处理。实施例1-24实施例1-24的实验细节(反应条件、试剂馈料、初始和最终催化剂浓度)、放大参数(相对进料和相对每小时进料速率)和结果(如通过gpc确定的聚合物分子量分布[mwd]和聚合物收率)以表格形式呈现在表iii-vi中。应该清楚的是,本发明的loxsh催化剂和聚合反应条件提供合成它们的试剂的无数组合和进行该方法的反应参数。因此,实施例1-5和实施例6-13包括设计用于探究该新型催化剂体系和反应条件的初始范围确定实验。因此,这些实施例每次实验仅利用约25%至50%的优选单体量。在采用的苯乙烯单体量降低的情况下,采用叶轮构造ii。因此,除了在聚合反应中放置叶轮和缩短苯乙烯单体进料外,这些实验的准备和实施基本上与实施例14-24相同。因此,实施例3、14-15、20-21和24被认为是代表性的并且更详细地描述。应该注意的是,由此生成的loxlihps组合物(实施例1-24)的四至六种最易挥发的苯乙烯低聚物的gc分析证明,99.4%至99.9%的离散聚合物链具有纯线性“头对尾”聚合物微结构。另外,这些loxlihps组合物基本上不含任何断裂聚合杂质或共产物分布。基于各种单体重复单元,这些分布的统计模型(weibullpdf)表明在99.986摩尔%和99.999摩尔%之间的苯乙烯重复单元具有2°(亚甲基)或3°(次甲基)苄基碳原子。换句话说,这些loxlihps组合物在微结构中具有小于10ppm至不大于140ppm的季碳“尾对头对尾”键合。实施例3代表loxlih约[dmea‾]5li12h7催化剂在80℃下25%的全单体进料体积在25℃下在干燥氢气(0psigh2)气氛下,将495ml(384.9g)无水环己烷中的345ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型ii的三节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由2.27g(0.0255摩尔)n,n-二甲基乙醇胺、134.7g(1.27摩尔)乙苯和12.22g(0.105摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水环己烷冲洗。接下来,将30.37ml(0.0607摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水环己烷等分试样。在有机锂馈料期间,搅拌速度增加至1130rpm,因此在15分钟馈料期过程中,随着氢气被消耗,反应器压力降至-3psig。将反应器顶部空间吹扫并用21psig干燥h2(经表面下进料管线)放空两次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在0psig下。然后将反应器加热至70℃,压力增长到4psig。用在反应器夹套上的81℃油进行加热。达到70℃后,开始苯乙烯单体进料,进料了257.2gg(2.47摩尔)的苯乙烯。经38分钟的时间,相对于6psig的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端,2.02ft/s)进料,控制反应温度在80℃下。在开始单体进料的10分钟内,反应器温度达到79℃并且压力增加至13psig。通过关闭调节器的阀门并测定下降5psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水乙苯冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水和200ml环己烷的洗涤容器中。在转移未猝灭的反应混合物期间,获得10ml样品用于分析。样品基本上是水白色的-即无色且透光-没有沉降或悬浮的固体。通过从移液管中添加一滴甲醇来猝灭样品。甲醇猝灭非常缓慢地产生氢气,但在超过1小时的时间内恒定地生成氢气-氢气似乎是从仅在初始猝灭时形成的极小颗粒中释放出来的。因此,由此形成的催化剂令人惊讶地非常缓慢地在聚合的终结条件下在环己烷中进行甲醇分解。包含二聚物内容物的粗猝灭反应混合物的gpc分析如下:mn:702,mw:1091,mz:1489,pd:1.554,σn=523,nα3=1.537-因此,这些条件生成不对称性异常低的线性(没有分支)阴离子链转移分布。标准后处理和产物分离将两相产物混合物在洗涤反应器中加热至65℃,然后分离各相。在65℃时容易地进行相切,并且其迅速需要很少的沉降时间。通过底部排液阀除去水和任何碎片或乳液。监测从反应器中除去的洗涤水的ph,第一洗涤液的ph值总是(意味着所有先前和随后的类似运行)等于14。进行另外的脱氧水洗涤;除去的水洗相的ph值为约12。然后用300ml3重量%h2so4洗涤有机相,接着用两份300ml自来水洗涤,最终ph为7。水洗产物混合物在环己烷和乙苯的洗涤反应器中通过常规蒸馏,同时将洗涤反应器的夹套温度逐渐降低到165℃来汽提。当釜温达到140℃以上时,认为蒸馏完成。使溶液冷却,然后收集394.75g的溶液。然后使用刮膜蒸发器(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)将该溶液进一步汽提出乙苯。该第一wfe操作生成245.0g具有如下包含二聚物的gpcmwd的loxlihps分布,mn:702,mw:1091,mz:1489,pd:1.554,σn=523,nα3=1.537。第二wfe操作(0.1-0.3mmhg真空,172.5℃,刮擦器速度为全速的60%,以1.0升/小时进料)提供217.9g具有2.40gpc面积%苯乙烯二聚物含量和如下gpcmwd的loxlihps分布,mn:821,mw:1152,mz:1505,pd:1.403,σn=521,nα3=1.417。实施例14和15使用低聚物微结构分析的情况下代表loxlih[dmea‾]4li6h2催化剂在80℃下全刻度单体进料体积在37℃下在干燥氢气(10psigh2)气氛下,将300ml(233.7g)无水环己烷中的150ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型iii的三节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由2.72g(0.0305摩尔)n,n-二甲基乙醇胺、140.0g(1.32摩尔)乙苯和1.82g(0.0157摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水环己烷冲洗。接下来,将22.90ml(0.0458摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水环己烷等分试样。在有机锂馈料期间,搅拌速度增加至1130rpm,并且在15分钟馈料期过程中,反应器压力降至9psig。将反应器顶部空间用干燥h2(经表面下进料管线)在50psig至0psig下吹扫并放空三次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在45psig下。然后将反应器加热至74℃,压力增长到63psig。用流经反应器夹套的81℃油进行加热。反应温度达到73℃后,开始苯乙烯单体进料,进料了960.4gg(9.22摩尔)的苯乙烯。经151分钟的时间,相对于的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端,1.88ft/s)进料,控制反应温度在80℃下。在开始单体进料的10分钟内,反应器温度达到81℃并且压力降到13psig。设定氢气调节器以维持16的psig的压力。通过关闭调节器的阀门并测定下降5psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计,氢吸收似乎是恒定或接近恒定的。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水乙苯冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水的洗涤容器中。因此,在洗涤反应器中小心地猝灭反应混合物。如实施例15在相同的馈料和条件下在按量配给试剂并重现这些条件的最小运行间变化内重复上述过程,不同的是经160分钟的时间进料1020.4gg(9.80摩尔)苯乙烯。如上所述,在添加进料的9分钟期间,氢吸收并且一定程度上降低。在转移未猝灭的反应混合物(实施例14和15)期间,获得10ml的各种反应混合物的样品用于分析。样品基本上是水白色的-即无色且透光-没有沉降或悬浮的固体。通过从移液管中添加一滴甲醇来猝灭样品。甲醇立即导致氢气的形成和释放。包含二聚物内容物的粗猝灭反应混合物的gpc分析如下:实施例14mn:439,mw:628,mz:886,pd:1.411,σn=288,nα3=2.108;实施例15mn:428,mw:636,mz:979,pd:1.539,σn=298,nα3=2.778。上述标准后处理提供3271g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)生成1793g具有如下包含二聚物的gpcmwd的loxlihps分布,mn:434,mw:630,mz:934,pd:1.452,σn=292,nα3=2.543。第二wfe操作(0.1-0.3mmhg真空,172.5℃,刮擦器速度为全速的60%,以1.0升/小时进料)提供1492g具有2.40gpc面积%苯乙烯二聚物含量和如下gpcmwd的loxlihps分布,mn:530,mw:731,mz:1150,pd:1.379,σn=326,nα3=3.661。执行第三wfe操作以获得低分子量低聚物,以确定loxlihps分布微结构。因此,将从第二wfe操作回收的1492g产物分布的163.2g样品汽提出低聚物(0.13mmhg真空,199.5℃,刮擦器速度为全速的85%,以2.0g/min进料)。该第三wfe操作生成31.24g具有如下gpcmwd的苯乙烯低聚物混合物,mn:310,mw:323,mz:337,pd:1.043。gc分析表明,99.940%的链具有期望的“头对尾”微结构,仅痕量(如果有的话)的链具有断裂的(fwi-14)微结构(参见图3)。应注意,实施例14和15几乎相同,不同之处在于在实施例15中采用另外量的苯乙烯单体。这些实施例利用叶轮构造iii,其对于实施例14的向凝相的氢转移是足够均匀的,但是据证明对于在实施例15中增加的60克单体进料刚好不够完全。与实施例14(表v)相比,这反映在实施例15的mwd的pdn、σn和nα3的值增加。因此,为了将更多单体进料到相同量的起始反应介质中,将第四叶轮加到如上面对于构造iv所指示的间隔开的搅拌轴中。在实施例20-21和24中利用构造iv。实施例21与实施例20相同,不同之处在于实施例21仅利用25%的优选总单体进料。这样做是为了探究分子量分布在运行过程中如何演变。基于该实验,pdn和nα3的值随着分子量的增加而减小,表明随着进料的继续,随着苯乙烯单体的每次增量形成宽度和不对称性差的分布,全都同时形成无活性聚合物链的递增不同的统计分布并且同时重整loxlih催化剂。这种实验组合将表明,可利用在稳态条件下操作的连续方法来形成具有期望的低多分散性、宽度和不对称性的甚至更优选的分子量分布。实施例20和21代表loxlih[dmea‾]2li6h4催化剂在80℃下全刻度单体进料体积在-5℃下在干燥氢气(12psigh2)气氛下,将300ml(231.0g)无水甲基环己烷中的150ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型iv的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由1.00g(0.0112摩尔)n,n-二甲基乙醇胺、140.0g(1.32摩尔)乙苯和2.60g(0.022摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水甲基环己烷冲洗。接下来,将16.81ml(0.0336摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水甲基环己烷等分试样。在有机锂馈料期间,搅拌速度增加至1130rpm,并且在15分钟馈料期过程中,反应器压力增加至14psig。将反应器顶部空间用干燥h2(经表面下进料管线)在50psig下吹扫并且放空三次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在40psig下。等到反应器温度达到18℃后,则将反应器加热到70℃,压力仅增长2psig,达到42psig,表明在加热后氢吸收。等到反应器达到72℃时,使h2压力增加到46psig,压力增长到60psig。用流经反应器夹套的81℃油进行加热过程。达到72℃后,开始苯乙烯单体进料,进料了1042.5g(10.01摩尔)的苯乙烯。经164分钟的时间,相对于的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端,1.88ft/s)进料,控制反应温度在80℃下。在开始单体进料的10分钟内,反应器温度达到80℃并且压力降到32psig。设定氢气调节器以维持14的psig的压力。通过关闭调节器的阀门并测定下降4psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计,氢吸收似乎是恒定或接近恒定的。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水乙苯冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水的洗涤容器中。因此,在洗涤反应器中小心地猝灭反应混合物。如实施例21在相同的馈料和条件下在按量配给试剂并重现这些条件的最小运行间变化内重复上述过程,不同的是经40分钟的时间进料255.0g(2.45摩尔)苯乙烯。在转移未猝灭的反应混合物(实施例20和21)期间,获得10ml的各种反应混合物的样品用于分析。样品基本上是水白色的-即无色且透光-没有沉降或悬浮的固体。通过从移液管中添加一滴甲醇来猝灭样品。甲醇立即导致氢气的形成和释放。包含二聚物内容物的粗猝灭反应混合物的gpc分析如下:实施例20mn:466,mw:675,mz:951,pd:1.409,σn=312,nα3=2.033;实施例21mn:408,mw:598,mz:932,pd:1.559,σn=278,nα3=2.993。上述但在82℃下进行的标准后处理提供1631.5g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)产生1168gloxlihps分布,其具有如下包含二聚物的gpcmwdmn:424,mw:626,mz:979,pd:1.476,σn=293,nα3=2.9794。第二wfe操作(0.1-0.3mmhg真空,172.5℃,刮擦器速度为全速的60%,以1.0升/小时进料)提供946g具有1.40gpc面积%苯乙烯二聚物含量和如下gpcmwd的loxlihps分布,mn:536,mw:722,mz:1049,pd:1.379,σn=326,nα3=2.912。实施例24使用低聚物微结构分析的情况下代表loxlih[dmea‾]5li12h7催化剂在80℃下全刻度单体进料体积在-5℃下在干燥氢气(13psigh2)气氛下,将300ml(231.0g)无水甲基环己烷中的150ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型iv的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由2.27g(0.0255摩尔)n,n-二甲基乙醇胺、140.0g(1.32摩尔)乙苯和12.40g(0.107摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水甲基环己烷冲洗。接下来,将30.79ml(0.0616摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水甲基环己烷等分试样。在有机锂馈料期间,搅拌速度增加至1130rpm,并且在15分钟馈料期过程中,反应器压力增加至14psig。将反应器顶部空间用干燥h2(经表面下进料管线)在50psig至0psig下吹扫并放空三次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在41psig下。等到反应器温度达到72℃时,则将反应器加热到73℃,压力增长到62psig。用流经反应器夹套的81℃油进行加热过程。达到73℃后,开始苯乙烯单体进料,进料了1041.0g(10.00摩尔)的苯乙烯。经164分钟的时间,相对于的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端,1.88ft/s)进料,控制反应温度在80℃下。在开始单体进料的10分钟内,反应器温度达到81℃并且压力降到34psig。设定氢气调节器以维持14的psig的压力。通过关闭调节器的阀门并测定下降4psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计,氢吸收似乎是恒定或接近恒定的。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(酸性氧化铝),用50ml无水乙苯冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水的洗涤容器中。因此,在洗涤反应器中小心地猝灭反应混合物。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品用于分析。样品基本上是水白色的-即无色且透光-没有沉降或悬浮的固体。通过从移液管中添加一滴甲醇来猝灭样品。甲醇猝灭立即导致氢气的形成和逸出。包含二聚物内容物的粗猝灭反应混合物的gpc分析如下:mn:466,mw:675,mz:951,pd:1.409,σn=312,nα3=2.033。上述但在82℃下进行的标准后处理提供1312.9g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)产生966.2gloxlihps分布,其具有如下包含二聚物的gpcmwdmn:477,mw:685,mz:961,pd:1.436,σn=315,nα3=2.032。第二wfe操作(0.1-0.3mmhg真空,172.5℃,刮擦器速度为全速的60%,以1.0升/小时进料)提供828.4g具有1.2gpc面积%苯乙烯二聚物含量和如下gpcmwd的loxlihps分布,mn:575,mw:753,mz:933,pd:1.310,σn=320,nα3=1.933。执行第三wfe操作以获得低分子量低聚物,以确定loxlihps分布微结构。因此,将从第二wfe操作回收的966.2g产物分布的106.2g样品汽提出低聚物(<0.1mmhg真空,199.5℃,刮擦器速度为全速的85%,以2.0g/min进料)。该第三wfe操作生成33.17g具有如下gpcmwd的苯乙烯低聚物混合物,mn:372,mw:398,mz:426,pd:1.069。gc分析表明,99.82%的链具有期望的“头对尾”微结构,仅痕量(如果有的话)的链具有断裂的(fwi-14)微结构(参见图4)。在完成系列的loxlih运行(实施例29-35)后,将高压釜反应器用标准鼓级(非无水)环己烷漂洗,用氮气充分吹扫,然后打开检查。用于所有意图和目的的加热的反应器壁和冷表面(即,冷却盘管、搅拌器组件、浸入管、单体进料管线和热电偶套管)不含所有固体。实施例25-29实施例25-29的实验细节(反应条件、试剂馈料以及初始和最终催化剂浓度)、放大参数(相对进料和相对每小时进料速率)和结果(如通过gpc确定的聚合物分子量分布和聚合物收率)以表格形式呈现在表vii中。如上所述,这里重申,应该清楚的是本发明的loxsh催化剂和聚合反应条件提供合成它们的试剂的无数组合和进行该方法的反应参数。这些实施例25-29包括涉及其他类盐氢化物的双金属催化剂。因此,实施例25-27需要使用氢化钾来形成loxkh催化剂,并且实施例28-29需要使用有机镁试剂形成loxmgh2催化剂。更详细地描述实施例26、28和29。应该注意的是,由此生成的loxmgh2ps组合物(实施例28-29)的四至六种最易挥发的苯乙烯低聚物的gc分析证明,99.2%的基本上不含任何断裂聚合杂质或共产物分布的线性“头对尾”聚合物微结构。这些分布的统计模型(betapdf)表明约99.982摩尔%的苯乙烯重复单元具有2°(亚甲基)或3°(次甲基)苄基碳原子。换句话说,这些loxmgh2ps组合物具有不超过185ppm的季碳“尾对头对尾”键合。loxkh生成的低聚物的分析显示非常高的水平-8%至12%的离散聚合物链-的组合物具有单个季碳头部“尾对头对尾”键合。因此,基于这些组合物的统计模型(betapdf),loxkh组合物可具有小于99.725摩尔%的具有2°(亚甲基)或3°(次甲基)苄基碳原子的苯乙烯重复单元。这意味着这些组合物可具有大于2750ppm的季碳“尾对头对尾”键合,这使得它们对于某些应用而言是不太优选的组合物。令人惊奇的是,甚至由li:k比为15:1的loxkh催化剂生成的实施例27的组合物也生成具有大于8重量%的具有不期望的单个季碳“尾对头对尾”键合的链的组合物。部分地基于表vii中呈现的结果假设,由一份dmeah与2份第i族金属(m+)形成的催化剂作为具有式[dmea-]4m8h4的聚集体存在。基于该假设,实施例27的催化剂(15:1li:k)可由1个具有式[dmea-]4li8h4的聚集体和一个具有式[dmea-]4li7kh4的聚集体组成。实施例25的催化剂(3:1li:k)可以完全是一个或多个具有式[dmea-]4li6k2h4的聚集体,并且实施例26的催化剂(7:1li:k)可以是具有式[dmea-]4li7kh4的聚集体。其中对于这些聚集体催化剂体系中的每一种,最具活性的氢化物物质就引发而言且因此就形成不太期望的聚合物微结构而言是kh,因为这三个实施例(25、26和27)提供与所有其他一样的基于钾的催化剂体系。与以loxmgh2聚集体催化剂体系为代表的实施例28和29的loxkh相比并形成对比,看起来loxlih聚集体催化剂的活性比loxmgh2大。对于实施例28,dmeah与正丁基锂与二丁基镁的化学计量比使得如果形成单个聚集体,则其将具有经验式[dmea-]21li28mg4h15,因此应该或至少可以存在lih活性物质和mgh2活性物质。预计形成几种不同的聚集体,其中一些可能不含镁,因此作为活性试剂的lih形式将存在于催化剂组合物中。相比之下,实施例29的正丁基锂和二丁基镁馈料达到这样的程度以致消耗所有的锂烷基基团(丁基锂基团),而仅留下烷基镁基团(二丁基镁基团)。实施例29的化学计量并预期的经验式为[dmea-]4li4mgh2,因此将不存在明确形式的lih。由于实施例28可以在比实施例29显著更低的氢气压力下运行以获得类似的产物mwd,所以据推测可能存在所有的锂催化剂聚集体并且其在作为用于氢介导的类盐氢化物引发的聚合过程的催化剂方面比包含一定量的氢化镁的催化剂聚集体更具活性。同样,形成实施例29催化剂,使得将不存在活性lih(即,dmeah的摩尔数超过馈入的有机锂的摩尔数)。该催化剂体系非常有效,但需要比实施例28高得多的h2压力。鉴于商用反应器的需求(维持恒定压力和适当密封所需的背压的简单量方面的挑战),对于适度高的h2压力的这种需要被认为是优于loxlih催化剂(实施例1-24))和实施例28的混合loxlihloxmgh2催化剂的偶然优势。实施例25-27的“[dmea‾]2lik•2tmeda储备溶液的形成与被2摩尔至5摩尔的tmeda容易地溶解于烃溶剂中的叔丁醇钾不同,由n,n-二甲基乙醇胺[dmea-]k形成的醇钾似乎即使在添加tmeda的情况下溶解性也差。然而,由氢化钾、正丁基锂和dmeah形成的混合金属醇盐容易地溶解于乙苯中。因此,在氮气氛下,在182.5(1.72摩尔无水乙苯中由1.10g(0.274)干燥的新鲜kh、4.90g(0.0550摩尔)dmeah、6.51(0.560摩尔)tmeda和15.12ml(0.0302摩尔)2.0m在环己烷中的正丁基锂制备6.12重量%的均匀的206.71gtmeda络合的混合金属氨基醇盐“[dmea-]2lik∙2tmeda”乙苯溶液的储备溶液。这通过在氮气吹扫的杂物箱中将3.67g30重量%在矿物油中的kh馈入放置到搅拌器加热板上的预先称重的烘箱干燥的500ml硼硅酸盐玻璃瓶和玻璃涂覆的搅拌棒来实现。然后洗涤氢化钾悬浮液并用30ml的无水正戊烷倾析三次。在干燥氮气流下干燥至恒重后,然后馈入182.46g乙苯、6.51gtmeda(/99.5%)和4.90gdmeah(/99.5%,按放出的h2的份数添加)。将所得异质溶液温和地加热至50℃并缓慢添加正丁基锂直至形成均匀溶液。需要10摩尔%过量的正丁基锂来生成具有持久微弱红色的均匀溶液,表明存在其他质子物质。添加一滴dmeah以猝灭红色,并且在冷却至室温后,储备溶液保持均匀。然后将该6.12重量%的含有期望量的钾离子的均匀储备溶液的等分试样用于形成实施例25-27的反应混合物。实施例26使用低聚物微结构分析的情况下代表在80℃下loxkh[dmea‾]4li7kh4催化剂在37℃下在干燥氢气(10psigh2)气氛下,将300ml(233.7g)无水环己烷中的150ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型iii的三节距叶片涡轮机)中通过正氮气压力经馈料容器馈入51.68g上述6.12重量%均匀溶液,其中该溶液是与溶解于50.0ml无水环己烷中的另外1.23g(0.0138摩尔)n,n-二甲基乙醇胺和1.63g(0.0140摩尔)的tmeda组合。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水环己烷冲洗。接下来,将用42.71g(0.403)无水eb稀释的20.66ml(0.0413摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水环己烷等分试样。在有机锂馈料期间,搅拌速度增加至1130rpm,并且在18分钟馈料期过程中,反应器压力降至8psig。将反应器顶部空间用干燥h2(经表面下进料管线)在50psig下吹扫并放空三次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在41psig下。然后将反应器加热至75℃,压力增长到62psig。用在反应器夹套上的81℃油进行加热。达到75℃后,开始苯乙烯单体进料,进料了996.9g(9.57摩尔)的苯乙烯。经156分钟的时间,相对于的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端,1.88ft/s)进料,控制反应温度在81.5℃下。在开始单体进料的10分钟内,反应器温度达到82℃并且压力降到47psig。设定氢气调节器以维持22的psig的压力。通过关闭调节器的阀门并测定下降5psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计,氢吸收在单体进料过程期间有所下降,这表明除了氢之外乙苯表现为该催化剂组合物的链转移剂。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水乙苯冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水的洗涤容器中。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品用于分析。样品是红色的并且透光,没有沉降或悬浮的固体。通过从移液管中添加一滴甲醇来猝灭样品。甲醇猝灭立即导致氢气的形成和逸出。基于包含二聚物内容物的粗猝灭反应混合物的gpc分析如下:mn:688,mw:1051,mz:1461,pd:1.527,σn=500,nα3=1.725。上述但在82℃下进行的标准后处理提供1159.5g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)生成935gloxkhps分布,其具有如下包括二聚物的gpcmwdmn:689,mw:1051,mz:1461,pd:1.527,σn=500,nα3=1.725。第二wfe操作(0.1至0.3mmhg真空,172.5℃,刮擦器速度为全速的60%,以1.0升/小时进料)提供845g具有0.48gpc面积%苯乙烯二聚物含量和如下gpcmwd的loxkhps分布,mn:765,mw:1099,mz:1486,pd:1.437,σn=505,nα3=1.667。执行第三wfe操作以获得低分子量低聚物,以确定loxkhps分布微结构。因此,将从第二wfe操作回收的845g产物分布的150.2g样品汽提出低聚物(<0.1mmhg真空,199.5℃,刮擦器速度为全速的85%,以2.0g/min进料)。该第三wfe操作生成18.94g具有如下gpcmwd的苯乙烯低聚物混合物,mn:34,mw:371,mz:400,pd:1.077。gc分析表明,89.53%的链具有期望的“头对尾”微结构,0.78%的链具有断裂(fwi-14)微结构,并且余量的链在离散聚合物链中具有不太期望的单一季“尾对头对尾”键合(参见图6)。实施例28代表loxmgh2[dmea‾]21li28mg4h15催化剂在80℃下的全单体进料在-5℃下在干燥氢气(13psigh2)气氛下,将375ml(288.8g)无水甲基环己烷中的175ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型vi的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由3.32g(0.0372摩尔)n,n-二甲基乙醇胺、30.0g(0.28摩尔)乙苯和6.51g(0.056摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水甲基环己烷冲洗。接下来,将溶解在80g(0.75摩尔)环己烷中的24.50ml(0.0490摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水甲基环己烷等分试样。然后,将溶解在30.0g(0.28摩尔)乙苯中的7.00ml的1.0m在庚烷中的二丁基镁(0.007摩尔)经馈料容器馈料并转移到反应器中,然后加入来自上述总量的50ml无水甲基环己烷等分试样。在有机锂/有机镁馈料期间,搅拌速度增加至1130rpm,并且在15分钟馈料期过程中,反应器压力增加至16psig。将反应器顶部空间用干燥h2(经表面下进料管线)在50psig下吹扫并且放空三次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在40psig和-3.4℃下。然后经一定时间(45分钟)将反应器加热到40℃。反应器温度达到40℃,压力增长到55psig。将反应器放空至46psig并继续加热。再加热15分钟后,反应器达到67℃,并且压力设定为63psig。用在反应器夹套上的81℃油进行加热过程。达到72℃和64psig后,开始苯乙烯单体进料,进料了1009.0g(9.69摩尔)的苯乙烯。经159分钟的时间,相对于的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端,1.88ft/s)进料,控制反应温度在82℃下。在开始单体进料的10分钟内,反应器温度达到80℃并且压力降到36psig。设定氢气调节器以维持14psig的压力,持续接下来的40分钟进料时间。在进料单体总共60分钟后,将氢气压力设定到11psig。通过关闭调节器的阀门并测定下降4psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计的氢吸收似乎是接近恒定的。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(酸性氧化铝),用50ml无水乙苯冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml的3重量%h2so4的洗涤容器中。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品用于分析。样品为浅黄色,没有观察到固体。通过从移液管中添加一滴甲醇来猝灭样品。甲醇猝灭立即导致黄色猝灭以及氢气的形成和逸出。包含二聚物内容物的粗猝灭反应混合物的gpc分析如下:mn:504,mw:773,mz:1180,pd:1.534,σn=368,nα3=2.538。将两相产物混合物在洗涤反应器中加热至82℃,然后分离各相。在82℃时容易地进行相切,并且其迅速需要很少的沉降时间。然后用4×300ml的自来水洗涤有机相,直至达到最终ph7。来自上述标准产物分离的标准溶剂汽提提供1561.8g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)生成962g具有如下包含二聚物的gpcmwd的loxmgh2ps分布,mn:511,mw:780,mz:1187,pd:1.526,σn=371,nα3=2.530。第二wfe操作(0.1-0.3mmhg真空,172.5℃,刮擦器速度为全速的60%,以1.0升/小时进料)提供828.4g具有1.1gpc面积%苯乙烯二聚物含量和如下gpcmwd的loxmgh2ps分布,mn:622,mw:853,mz:1207,pd:1.371,σn=379,nα3=2.417。实施例29使用低聚物微结构分析的情况下代表loxmgh2[dmea‾]4li4mgh2催化剂在80℃下50%单体进料体积在-5℃下在干燥氢气(12psigh2)气氛下,将375ml(288.8g)无水甲基环己烷中的175ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型vi的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由5.00g(0.0561摩尔)n,n-二甲基乙醇胺、30.0g(0.28摩尔)乙苯和8.15g(0.056摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水甲基环己烷冲洗。接下来,将溶解在80g(0.75摩尔)环己烷中的28.05ml(0.0561摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水甲基环己烷等分试样。然后,将溶解在30.0g(0.28摩尔)乙苯中的14.00ml的1.0m在庚烷中的二丁基镁(0.014摩尔)经馈料容器馈料并转移到反应器中,然后加入来自上述总量的50ml无水甲基环己烷等分试样。在有机锂/有机镁馈料期间,搅拌速度增加至1130rpm,并且在16分钟馈料期过程中,反应器压力增加至17psig。将反应器顶部空间放空到0psig,然后用干燥h2(经表面下进料管线)加压到65psig并放空三次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在44psig和-4.4℃下。然后经120分钟将反应器加热到71℃,反应过程用在反应器加热套上的81℃油进行。达到71℃和61psig后,开始苯乙烯单体进料,进料了509.0g(4.89摩尔)的苯乙烯。经80分钟的时间,相对于的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端,1.88ft/s)进料,控制反应温度在81℃下。在开始单体进料的10分钟内,反应器温度达到80℃并且压力降到49psig。(然而,需要指出的是,与loxlih运行相比,如压力下降所证明的氢消耗延迟约0.25分钟至0.75分钟。)设定氢气调节器以维持46psig的压力,持续接下来的40分钟进料时间。在进料单体总共60分钟后,将氢气压力控制在65psig下。通过关闭调节器的阀门并测定下降4psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计的氢吸收似乎是接近恒定的。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水乙苯冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml的3重量%h2so4的洗涤容器中。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品用于分析。样品为浅黄色,一些大粒子沉降。通过从移液管中添加一滴甲醇来猝灭样品。甲醇猝灭立即导致黄色的猝灭以及氢气的形成和逸出。包含二聚物内容物的粗猝灭反应混合物的gpc分析如下:mn:481,mw:713,mz:1008,pd:1.482,σn=334,nα3=1.969。实施例28的处理和汽提程序提供729.9g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)生成492g具有如下包含二聚物的gpcmwd的loxmgh2ps分布,mn:485,mw:718,mz:1018,pd:1.480,σn=336,nα3=2.000。第二wfe操作(0.1-0.3mmhg真空,172.5℃,刮擦器速度为全速的60%,以1.0升/小时进料)提供427g具有1.1gpc面积%苯乙烯二聚物含量和如下gpcmwd的loxmgh2ps分布,mn:593,mw:805,mz:1116,pd:1.358,σn=355,nα3=2.256。执行第三wfe操作以获得低分子量低聚物,以确定loxmgh2ps分布微结构。因此,将从第二wfe操作回收的492g产物分布的94.5g样品汽提出低聚物(0.12mmhg真空,199.5℃,刮擦器速度为全速的85%,以2.0g/min进料)。该第三wfe操作生成15.73g具有如下gpcmwd的苯乙烯低聚物混合物,mn:314,mw:329,mz:344,pd:1.049。gc分析表明,99.21%的链具有期望的“头对尾”微结构,不含具有断裂的(fwi-14)微结构的链,其中痕量的低聚物用丁基基团引发(参见图8)。观察到的氢消耗的轻微延迟和用丁基引发的痕量聚苯乙烯分布的存在可以提示在通过催化剂组合物中残留丁基镁基团使苯乙烯烷基化之后,更完全地实现了氢化镁组合物的形成。这可以提示苄基镁或苄基菱镁矿试剂比脂族镁或脂族菱镁矿试剂更容易被氢还原。完成该系列loxmgh2运行(实施例28和29)后,用标准鼓级(非无水)环己烷漂洗高压釜反应器,用氮气充分吹扫,然后打开检查。加热的反应器壁和冷表面(即,冷却盘管、搅拌器组件、浸入管、单体进料管线和热电偶套管)被鳞片状白色固体覆盖。从反应器表面漂洗掉固体的自来水冲洗液生成ph=11的漂洗液。分析(icp)表明,鳞片状固体由锂盐和镁盐组成:84.0毫克lioh(3.51毫摩尔的li或占总锂的3.34%)和0.24毫克(0.004毫摩尔的mg或占总镁的0.02%)mg(oh)2。在这两次运行之间,已经馈入105.1毫摩尔的锂和21.02毫摩尔的镁,因此固体残留物仅代表小百分数的锂并且基本上是痕量的馈入的镁。实施例30-32实施例30-32的实验细节(反应条件、试剂馈料以及初始和最终催化剂浓度)、放大参数(相对进料和相对每小时进料速率)和结果(如通过gpc确定的聚合物分子量分布和聚合物收率)以表格形式呈现在表viii中。实施例30-32需要形成其他loxlih催化剂和方法,其中络合配体是甲氧基乙醇锂[meoe-]li+或2-n,n-二甲基氨基乙氧基乙醇锂[dmaeoe-]li+。实施例30证明,在氢气氛下,在tmeda存在下,可由2-甲氧基乙醇[meoeh]、正丁基锂形成具有[meoe-]4li8h4∙4tmeda的经验式的烃可溶性氢化锂试剂或催化剂。实施例30还证明该催化剂体系将引发苯乙烯单体的聚合,然而氢介导或链转移过程在该实施例的条件下是低效的,每摩尔络合的氢化锂仅生成0.86摩尔的聚合物链。这可能意味着如果催化剂促进或有利于涉及活跃的聚(苯乙烯基)锂物质的氢链转移反应,则必须在催化剂的锂醇盐络合剂上存在胺官能团。应注意,具有经验式[meoe-]4li12h8的烃可溶性氢化锂组合物对于烃可溶性形式的lih将具有非常高的氢化物含量:2.06重量%的氢化物。同样,具有经验式[meoe-]4li8h4的组合物将具有高氢化物含量-1.12重量%的氢化物。对于实施例31和32,使用2-n,n-二甲基氨基乙氧基乙醇[dmaeoeh]形成氢化物试剂或催化剂物质。实施例31中使用的试剂馈料使得将形成具有经验式[dmaeoe‾]4li8h4∙4tmeda的催化剂。然而,基于几乎没有氢吸收的证据,从一开始就清楚形成了很少的催化剂。据推测,tmeda有利于[dmaeoe‾]li+物质被正丁基锂分解。因此,该催化剂在该实施例的条件下生成非常高分子量(mw=183,233)的聚苯乙烯组合物,其具有非常宽且高度不对称的分布。据推测,tmeda有利于[dmaeoe‾]li+物质被正丁基锂分解。实施例32中使用的试剂馈料使得将形成不含tmeda的具有经验式[dmaeoe‾]4li6h2的催化剂。另外,催化剂初始在-5℃下形成。在实施例32的反应条件下,形成且至少初始形成的期望的催化剂具有与由dmaeh形成的催化剂相当的活性,然而,在苯乙烯单体进料期间催化剂活性急剧下降,导致形成高分子量尾部,如由mz=139,795,不对称性为34.4所表明。据推测,在反应的温度条件下导致催化剂分解。无论是在催化剂形成期间还是在聚合过程期间,[dmaeoe‾]li+的分解都被认为需要对二甲氨基官能团进行金属化α,然后消除乙二醇的二锂醇盐并形成如下所示的乙烯基-二甲胺(烯胺)。因此,该配体和易受这类可能的降解过程影响的任何其他配体对于用于形成用于涉及单金属锂催化剂的氢介导的类盐氢化物引发的聚合方法的催化剂的dmeah不太优选。实施例30代表由2-甲氧基乙醇形成的loxlih[meoe‾]4li8h4∙4tmeda催化剂在20℃下在干燥氢气(0psigh2)气氛下,将550ml(428.5g)无水环己烷中的400ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型iii的三节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由2.10g(0.0276摩尔)2-甲氧基乙醇、15.0g(0.14摩尔)乙苯和3.35g(0.029摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水环己烷冲洗。接下来,经15分钟将溶解在80g(0.75摩尔)环己烷中的28.77ml(0.0575摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水环己烷等分试样。在有机锂馈料期间,搅拌速度增加至1130rpm,并且反应器压力减小到-4psig且温度增加到22℃,指示h2的消耗。将反应器顶部空间用50psig至0psig干燥h2(经表面下进料管线)吹扫并放空三次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在35psig下。然后经60分钟将反应器加热至73℃,其中用流经反应器夹套的81℃油进行加热过程。达到73℃和51psig后,开始苯乙烯单体进料,进料了160.0g(1.54摩尔)的苯乙烯。经35分钟的时间,相对于的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端,1.35ft/s)进料,控制反应温度在80℃下。在开始单体进料的10分钟内,反应器温度达到78℃并且压力增加到52psig。氢气调节器的阀门保持关闭,并且压力随着顶部空间被苯乙烯单体进料压缩而增加。在进料总共35分钟后,氢气压力达到55psig。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水乙苯冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水的洗涤容器中。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品。样品是无色的并且透光,没有沉降或悬浮的固体。通过从移液管中添加一滴甲醇来猝灭样品。甲醇猝灭立即导致氢气的形成和逸出。当顶部空间的氮气喷射不能在顶部再吸收乙苯时认为除蒸馏之外的上述标准后处理和溶剂汽提完成。所得树脂经底部排液阀转移到预先称重的衬有铝箔的金属托盘中。然后在真空烘箱中将树脂进一步汽提出乙苯,该真空烘箱被从100℃逐渐加热至165℃,真空从50.0mmhg逐渐增加至1.0mmhg真空。冷却后,将所得脆性无色树脂取样并通过gpc分析:mn:6179,mw:14,450,mz:22,964,pd:1.578,σn=7192,nα3=2.338。实施例32代表由2-n,n-二甲基氨基乙氧基乙醇形成的loxlih[dmaeoe‾]4li6h2催化剂在-5℃下在干燥氢气(0psigh2)气氛下,将300ml(231.0g)无水甲基环己烷中的150ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型iii的三节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由4.00g(0.0300摩尔)2-n,n-二甲基氨基乙氧基乙醇和16.0g(0.14摩尔)乙苯形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水甲基环己烷冲洗。接下来,经15分钟将溶解在120g(1.13摩尔)环己烷中的22.54ml(0.0451摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水甲基环己烷等分试样。在有机锂馈料期间,搅拌速度增加至1130rpm,并且反应器压力既不增加也不减小。然后将反应器顶部空间用干燥h2(经表面下进料管线)加压到50psig并且放空三次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在36psig下。然后经60分钟将反应器加热至73℃,其中用在反应器夹套上的81℃油进行加热过程。达到72℃和59psig后,开始苯乙烯单体进料,进料了131.1g(1.26摩尔)的苯乙烯。经29分钟的时间,相对于的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端,1.35ft/s)进料,控制反应温度在80℃下。在开始单体进料的10分钟内,反应器温度达到82℃并且压力降到52psig。将氢气调节器设定到36psig。在苯乙烯单体进料的前10-15分钟期间,该方法似乎与由dmea形成的催化剂体系相当。然而,很明显,到20分钟,氢吸收迅速减少。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(酸性氧化铝),用50ml无水乙苯冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水的洗涤容器中。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品。样品是无色的并且透光,没有沉降或悬浮的固体。通过从移液管中添加一滴甲醇来猝灭样品。甲醇猝灭立即导致氢气的形成和逸出。当顶部空间的氮气喷射不能在顶部再吸收乙苯时认为除蒸馏之外的上述标准后处理和溶剂汽提完成。所得树脂经底部排液阀转移到预先称重的衬有铝箔的金属托盘中。然后在真空烘箱中将树脂进一步汽提出乙苯,该真空烘箱被从100℃逐渐加热至165℃,真空从50.0mmhg逐渐增加至1.0mmhg真空。冷却后,将所得脆性无色树脂取样并通过gpc分析:mn:745,mw:23,605,mz:139,795,pd:5.922,σn=4127,nα3=34.431。实施例33-37实施例33-37的实验细节(反应条件、试剂馈料以及初始和最终催化剂浓度)、放大参数(相对进料和相对每小时进料速率)和结果(如通过gpc确定的聚合物分子量分布和聚合物收率)也以表格形式呈现在表viii中。这些实施例(33-37)经设计是在烃气氛下进行,存在很少的氢气(如果有的话)。这些实施例的目的是证明以下四点:(1)本发明的loxlih催化剂引发单体如苯乙烯的聚合;(2)这些实施例的所得mn分子量为形成的loxlih催化剂聚集体的实际组成提供了一些实验证据;(3)证明添加的促进剂tmeda确实在确定催化剂活性方面起作用;和(4)由于在这些实施例中不存在氢,氢气清楚且明确地具有介导且甚至进一步活化本发明的类盐氢化物引发的聚合方法的惊人且令人惊讶的效果。因此,利用lih:苯乙烯比为1:31.9的经验式[dmea‾]2li3h的催化剂(mn计算=3316)的实施例33提供类盐氢化物引发的聚苯乙烯组合物,其中mn=6707,%效率≈50%。假设没有自乙苯链转移,这将意味着在聚集体中仅二分之一的lih可用以引发聚合,并且将表明实际的催化或引发物质具有化学式[dmea-]4li6h2。相比之下,采用相同催化剂体系但每摩尔锂金属存在1摩尔tmeda的实施例34生成类盐氢化物引发的聚苯乙烯组合物,其mn=5084。因此,该tmeda馈料似乎增加了氢化物对由2摩尔dmeah和3摩尔正丁基锂形成的催化剂的效率和可能的可用性。与实施例33和34形成对比,实施例35和36的催化剂体系具有令人惊讶的完全不同的行为。因此,实施例35和36利用具有经验式[dmea-]lih的催化剂(实施例35没有tmeda和实施例36有tmeda0.5摩尔tmeda/摩尔总锂)。如表viii中所示,实施例35和36的%效率分别为35%和26%。同样,假设乙苯很少或没有作为链转移剂参与,这将表明催化剂的形成分别具有[dmea-]3li6h3和[dmea-]4li8h4∙4tmeda的经验式或实际式的组成,其中在两种情况下仅一种氢化物可以加成到苯乙烯上并形成聚合物引发物质。从实施例33-36的催化剂的%效率:约50%、约66.7%、约33.3%和约0.25%可以显而易见,loxlih催化剂和引申开来本发明的loxsh催化剂根据在其形成中使用的试剂可能作为相对简单的限定化学计量的聚集体存在。然而,实施例37可以提示可以形成复杂得多但明确限定的超级聚集体。因此,利用lih:苯乙烯比为1:18.6的经验式[dmea‾]li3h2的催化剂(mn计算=1933)的实施例37提供类盐氢化物引发的聚苯乙烯组合物,其中mn=17,972,%效率≈11%。这将表明仅1/9的lih可用于加成到苯乙烯上并因此引发聚合。这可以表明聚集体的混合物具有比率为75:25的经验式[dmea-]8li16h8和[dmea-]6li18h12,其中1/8和1/12的lih可用于引发聚合。因此,尽管我们希望不受刚刚关于loxlih催化剂的实际化学式呈现的理论的约束,但实施例33-37清楚地证明了上述四点(1)-(4)。提供实施例37以代表实施例33-37。实施例37代表在烃气氛下在82℃下的loxlih[dmea‾]li3h2∙2tmeda在10℃下在干燥氢气(11psigh2)气氛下,将650ml(506.4g)无水环己烷中的450ml馈入反应器中。向搅拌的溶剂(800rpm,具有上述构型iii的三节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由溶解于50ml的上述总量的无水环己烷中的3.02g(0.0339摩尔)n,n-二甲基氨基乙醇和7.90g(0.068摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水环己烷冲洗。接下来,经15分钟将溶解在65g(0.61摩尔)环己烷中的50.90ml(0.1018摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的两份50ml无水环己烷等分试样。在有机锂馈料期间,搅拌速度增加至1130rpm,反应器压力减小到5psig并且温度增加到13℃。然后将反应器顶部空间用干燥h2(经表面下进料管线)加压到50psig,然后放空到0psig,重复总共三次(缓慢放空以防止内容物从反应器中发泡),使反应器保持在46psig下。然后经50分钟将反应器加热至70℃,其中用流经反应器夹套的85℃油进行加热过程。达到73℃和57psig后,将氢气氛排出到矿物质油鼓泡器(0psig)中。继续加热直至反应器温度达到82℃并且排出管线从冷凝环己烷蒸气开始变暖,从而吹扫剩余的氢气并形成烃气氛。关闭矿物质油鼓泡器的阀门并将搅拌降至794rpm并且开始苯乙烯单体进料,进料了131.3g(1.26摩尔)的苯乙烯。相对于烃气氛经30分钟将苯乙烯经表面下进料管线(0.02”内径尖端,1.29ft/s)进料,将反应温度控制在82℃下。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水乙苯冲洗。将反应器加压至65psigh2,在搅拌增加至1130rpm后,压力降至60psig。将如此氢猝灭的阴离子聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水的洗涤容器中。在转移未猝灭的氢化物反应混合物期间,获得10ml反应混合物样品。样品是淡淡的粉红色的并且透光,没有沉降或悬浮的固体。在样品瓶中夹带的空气的情况下摇动样品,猝灭淡淡的粉红色。通过从移液管中添加一滴甲醇来猝灭样品。甲醇猝灭立即导致氢气的形成和逸出。当顶部空间的氮气喷射不能在顶部再吸收乙苯时认为除蒸馏之外的上述标准后处理和溶剂汽提完成。所得树脂(154g)经底部排液阀转移到预先称重的衬有铝箔的金属托盘中。然后在真空烘箱中将树脂进一步汽提出乙苯,该真空烘箱被从100℃逐渐加热至165℃,真空从50.0mmhg逐渐增加至1.0mmhg真空。冷却后,将所得脆性无色树脂(128g)取样并通过gpc分析:mn:17,972,mw:37,183,mz:49,015,pd:1.318,σn=18,581,nα3=1.299。实施例38-40实施例38-40是涉及苯乙烯单体和其他形式的类盐氢化物作为催化剂的氢介导的类盐氢化物引发的聚合方法的实例。实施例38的超活性类盐氢化物(sash)催化剂是在tmeda存在下由丁基锂和叔丁醇制备。该催化剂最多是微溶的,因此生成分子量高的hmship分布。实施例39的sash催化剂是在tmeda存在下除丁基锂之外由叔丁醇钾制备。该催化剂在形成低分子量hmship分布方面非常有效,然而该sash催化剂方法在聚合物微结构中形成不期望的季“尾对头对尾”键合(与所有其他基于钾的阴离子链转移聚合反应一样)。实施例40利用高活性类盐氢化物(hash)催化剂,该催化剂通过在氢气氛下将苯乙烯单体进料到在thf中的钠钾合金分散体中而形成。基于生成获得的分子量分布所需的钠和钾的克-原子,该hash催化剂的效率相对较低。另外,hash催化剂不出意地提供了不同微结构,特别是断裂低聚物的聚合物分布的复杂混杂物。实施例38生成高分子量聚苯乙烯分布组合物的超活性氢化锂催化剂方法在20℃下在氢气氛(0psig)下,将300g无水乙苯馈入反应器中。向搅拌的溶剂(800rpm,双节距叶片叶轮,叶片放置构造iii)中经馈料容器馈入预先由3.62g(0.0489摩尔)叔丁醇、69.9g(0.66摩尔)乙苯和23.50g(0.202摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用50g份的乙苯冲洗。将搅拌增加至1130rpm,然后将溶解在100g乙苯中的54.10ml(0.11摩尔)的2.0m正丁基锂经馈料容器缓慢转移到反应器中。反应器温度升高5℃达到25℃,压力增加到2psig,然后降至-4psig,将丁基锂溶液和后续50g乙苯漂洗等分试样加入反应器中。将含有总共570g(5.4摩尔)乙苯的反应器加热至90℃。在催化剂组分馈料期间引入的痕量n2通过用干燥h2(通过顶部空间)加压至50psig进行吹扫并放空三次(缓慢放空以防止内容物从反应器中发泡)。将h2调节器初始设定到21psig。经116分钟的时间,相对于氢气顶部压力,462.2g(4.44摩尔)的苯乙烯经表面下进料管线(0.02”内径尖端,1.2ft/s)进料,控制反应温度在90℃下,并且使氢气压力缓慢增加到41psig。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱,用50ml无水环己烷冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。在运行过程期间,定期关闭氢气调节器的阀门,以验证苯乙烯进料期间的氢吸收。该反应确实吸收氢气,虽然非常缓慢。将反应混合物的未猝灭内容物转移到预先馈有300ml加热至65℃的脱氧水的洗涤容器(n2气氛)中,然后用脱氧水(3×300ml)洗涤。然后在分离水性猝灭时适当地丢弃该反应混合物。在转移未猝灭的反应混合物的过程期间,获得10ml未猝灭反应混合物的等分试样。该无色样品充满均匀悬浮的极细碎固体。用甲醇猝灭样品,导致自粘性混合物中立即产生并逸出氢气。使用标准较高分子量柱和聚苯乙烯标准物的样品的gpc分析如下:gpcmwd,mn:1030,mw:5635,mz:10,066,pd:5.47,σn=2178,nα3=4.13。实施例39使用低聚物微结构分析的情况下代表对于在乙苯中在中等温度70℃下sash催化剂运行的80%单体进料体积在20℃下在干燥氮气氛下将300g(2.83摩尔)无水乙苯中的200g馈入反应器中。向搅拌的溶剂(800rpm,双节距叶片叶轮,叶片放置构造i)中经馈料容器馈入预先由4.57g(0.0407摩尔)叔丁醇钾、44g(0.41摩尔)乙苯和20.83g(0.179摩尔)tmeda形成的溶液。将反应器的馈料容器和转移管线用上述300g乙苯的50g份冲洗。接下来,将20.34ml(0.0407摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述的50g乙苯等分试样。将反应器加热到65℃。然后将搅拌增加到1130rpm并且通过用干燥h2(通过顶部空间)加压至65psig对反应器吹扫n2并放空三次(缓慢放空以防止内容物从反应器中发泡)。将h2调节剂设定到11psig,并且经183分钟的时间,相对于氢气顶部压力,800g(7.68摩尔)的苯乙烯经表面下进料管线(0.01”内径尖端,5.2ft/s)进料,控制反应温度在70℃下。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱,用50ml无水环己烷冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水以及500ml自先前运行蒸馏的回收的环己烷的洗涤容器中。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品用于分析。样品呈红色并对光透明,使其外观类似于活跃的aps工艺样品的颜色。样品的外观完全不同于在不存在氢气氛的情况下进行的样品阴离子链转移聚合的特征性深黑红(黑樱桃)颜色。在催化剂组分在形成氢化物之前在n2下组合的这种sash催化剂样品通常可含有大(mm尺寸)催化剂粒子。通过添加一滴甲醇猝灭样品,该甲醇立即猝灭红色并导致氢气的立即形成和逸出。粗猝灭反应混合物的gpc分析如下:mn:367,mw:497,mz:695,pd:1.35,σn=218,nα3=2.38。上述标准后处理和汽提程序提供1303g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器全速的60%,以1.0升/小时进料)生成827.9g具有如下gpcmwd的sashps分布,mn:376,mw:508,mz:707,pd:1.35,σn=223,nα3=3.34。第二wfe操作(0.1至0.3mmhg真空,172.5℃,刮擦器全速的60%,以1.0升/小时进料)提供608.7具有0.99gpc面积%苯乙烯二聚物含量和如下gpcmwd的sashps分布,mn:486,mw:593,mz:750,pd:1.22,σn=228,nα3=2.15。执行第三wfe操作以获得低分子量低聚物,从而确定sashps分布微结构。因此,将从第二wfe操作回收的608.7g产物分布的180.2g样品汽提出低聚物(<0.1mmhg真空,199.5℃,刮擦器速度为全速的85%,以2.0g/min进料)。该第三wfe操作生成33.17g具有如下gpcmwd的苯乙烯低聚物混合物,mn:332,mw:348,mz:363,pd:1.048。gc分析表明,93.49%的链具有期望的“头对尾”微结构,仅0.11%的链具有断裂的(fwi-14)微结构(参见图7)。实施例40使用低聚物微结构分析的情况下高活性类盐氢化物催化剂氢介导的苯乙烯聚合阴离子链转移方法在美国专利5,777,162和5,866,720中描述的改进的2升高压釜反应器中进行。在干燥氮气氛下在20℃下将818g无水四氢呋喃和6.2g(0.183g-原子碱金属)钠钾合金(nak2)馈入反应器中。用氢气(3×70psig)对未搅拌的反应混合物吹扫氮气并加压至70psigh2。施用高速高剪切混合(1900rpm)并将208.0g(2.00摩尔)苯乙烯经73分钟(3.15ml/min)进料到反应混合物中。在苯乙烯单体进料期间,反应器压力维持在70psig至60psigh2之间。在进料完成后,将反应器排出h2,并用异丙醇小心地猝灭反应混合物。通过gpc分析猝灭的反应混合物的样品,并具有以下mwd:mn:591,mw:943,mz:1438,pd:1.60,σn=456,nα3=2.38。将反应物料转移到含有乙苯的折皱性洗涤反应器中,水洗并汽提出thf。在刮膜蒸发器wfe(2”玻璃popestill,石墨叶片,在300.0mmhg真空下操作,140℃,刮擦器全速的60%,以1.0升/小时速率进料)的进一步汽提生成191g具有如下gpcmwd的聚苯乙烯树脂:mn:603,mw:956,mz:1373,pd:1.58,σn=461,nα3=1.906。对来自上面的191g中的164g样品进行第二wfe操作(在0.4mmhg真空下,230℃,刮擦器全速的60%,以1.0升/小时速率进料),产生153.6g具有如下gpcmwd的树脂:mn:802,mw:1081,mz:1418,pd:1.35,σn=473,nα3=1.645。第二wfe操作提供18.2g的二聚物、三聚物、四聚物、五聚物和六聚物的复杂混合物。gc分析证明,该复杂混合物来自包括头对尾聚合、链异构化和链断裂聚合的反应途径(参见图5)。完成制备二聚物汽提的loxlih和loxmgh2氢介导的类盐氢化物引发聚合产物分布。这些组合物中的11种根据pct公告wo2010/127091(us8,802,787b2)的工艺技术溴化以形成溴化的阴离子链转移乙烯基芳族聚合物(br-actvap)。下面列出这些溴化聚合物的物理特性的平均值和标准偏差,以及通过组合然后汽提实施例14-15的产物分布形成的br-actvap的特性,该br-actvap为具有mw=731和pdn=1.38的二聚物汽提的组合物。这些组合物在高抗冲聚苯乙烯(hips)中作为聚合物阻燃剂进一步测试,并且发现提供阻燃(ul94v0,在1/8”和1/16”下)hips制剂,其具有优异的整体特性,包括颜色(yi)、悬臂梁式冲击(izod)、热变形温度和vicat软化温度由于实施例14和15的催化剂组合物由[dmea-]li3h2∙1.0tmeda组成,并且该组合物在溴化后提供总体上优异的特性,所以认为所述该特定loxlih催化剂和所得氢介导的阴离子聚苯乙烯(hmaps)的进一步开发是有正当理由的。实施例41-42实施例41-42进一步证明经设计进一步阐明[dmea-]xliyhz的复杂化学计量的实验。这些实施例在除dmeah:正丁基锂的馈料比外相同的条件下进行。这两个实施例的数字详情呈现在表ix中。实验细节在下文呈现。实施例41代表在烃气氛下在77-79℃下的[dmea‾]li3h2催化剂在37.6℃下在干燥氢气(20psigh2)气氛下,将总共500ml(385.3g)无水环己烷中的220ml馈入反应器中。向搅拌的溶剂(600rpm,具有上述构型iv的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由溶解在上述总量无水环己烷中的20ml中的5.18g(0.0581摩尔)n,n-二甲基氨基乙醇形成的溶液。将反应器的馈料容器和转移管线用来自上述总量的50ml份的无水环己烷冲洗。接下来,经20分钟将溶解在上述总共500ml中的120ml环己烷中的44.95ml(0.0899摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述总量的50ml无水环己烷等分试样。在有机锂馈料期间,搅拌速度维持在600rpm下,在反应器压力升到24psig之后使其减小到18psig,并且温度增加到39.9℃。然后将反应器顶部空间用干燥h2(经表面下进料管线)加压到53psig,将搅拌增加到1000rpm,经30分钟的时间将催化剂溶液加热到69.9℃。在加热过程期间,h2压力达到64psig。将反应器进一步加压至76psig并搅拌2.5小时,然后在73.2℃下放空至0psig。然后经30分钟将反应器加热至92℃,同时用流经反应器夹套的115℃油进行加热过程。在达到92℃和8psig后,当环己烷蒸气开始在顶部冷凝时,将氢气氛排放到矿物油鼓泡器中,达到4psig,从而形成烃气氛。关闭矿物油鼓泡器的阀门,将搅拌降低到1100rpm,并将反应器冷却至76.9℃和-2psig压力。将98.0g(0.94摩尔)苯乙烯单体与90g环己烷组合。经60分钟的时间(5.0ml/min.),相对于烃气氛,将苯乙烯/环己烷进料经表面下进料管线(0.02”内径尖端)进料,控制反应温度在不会升高到高于夹套上的79.4℃(80-85℃)油的反应温度下。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水环己烷冲洗。将反应器加压至65psigh2。将氢猝灭的阴离子聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水的洗涤容器中。在转移未猝灭的氢化物反应混合物期间,获得10ml反应混合物样品。样品是淡淡的粉红色的并且透光,没有沉降或悬浮的固体。在样品瓶中夹带的空气的情况下摇动样品,猝灭淡淡的粉红色。当顶部空间的氮气喷射不能在顶部再吸收乙苯时认为除蒸馏之外的上述标准后处理和溶剂汽提完成。所得树脂(90g)经底部排液阀转移(具有很大的困难并且借助于高温空气喷枪)到预先称重的衬有铝箔的金属托盘中。通过gpc分析冷却的树脂提供以下:mn:13,845,mw:38,933,mz:65,777,pd:2.812,σn=18,637,nα3=2.84。实施例42-51实施例42至51证明,当使用tmeda作为促进剂时在进料过程中在从约275±50ppmlih变化降至约80±20ppmlih的催化剂浓度下由较快的相对进料速率和稍微降低的氢气压力导致共产物乙苯和苯乙烯二聚物的形成降低(mn增加),通常带来改善的收率。这些实施例的细节和结果呈现在表x中。在运行这组实施例的过程中,发现在催化剂形成期间tmeda的存在可具有失活的影响。因此,对于实施例42-47,只有在氢气氛下将dmeah和正丁基锂组合后才将tmeda馈入反应器中,其中反应器将已含有来自前一次运行的一定量的热(除了实施例42,其中反应器已经预先清洁过)。因此,对于实施例42-47,mn通常随着进料速率增加和h2压力降低而增加。然而,在实施例48中,随着进料速率增加和采用减小的h2(9psig)压力,mn降至457道尔顿。运行实施例47和48已经过了大于2周的时间,因此在此期间,任何留在反应器表面上的含有tmeda的反应混合物都已经流到反应器的底部。另外,因为反应器已经放置了这么长的时间,所以在形成实施例48的催化剂之前,使用500ml的无水环己烷冲洗来吹扫实施例47的任何剩余物。因此,在实施例49中,在馈入正丁基锂之前将tmeda馈入反应器中,结果该实施例尽管使用增加的h2(11psig)压力也生成具有mn=540的hmaps分布。因此,在后续两次运行实施例50和51中,对充分漂洗的反应器进行催化剂组分馈料。据推测,tmeda促进形成超活性但不溶的氢化锂,其将直接由正丁基锂、tmeda和氢气形成,而没有提供烃可溶性形式的lih的中间体dmeah。因此,实施例50和51一起代表表x的实施例,并且被认为是该表的实施例的优选方法的代表。实施例50和51代表[dmea‾]4li6h2•2tmeda催化剂在80℃下的全刻度单体进料体积在37℃下在干燥氢气(16psigh2)气氛下,将300ml(233.7g)无水环己烷中的150ml馈入充分漂洗的反应器中。向搅拌的溶剂(800rpm,具有上述构型iv的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由2.53g(0.0284摩尔)n,n-二甲基乙醇胺、总共170.0g(1.60摩尔)乙苯中的20g和上述300ml中的50ml形成的溶液。接下来,将溶解在120g无水乙苯(来自上述170g)和来自上述总量的无水环己烷的另一50ml等分试样中的21.25ml(0.0425摩尔)2.0m正丁基锂在馈料容器中组合,然后在氢气下经15分钟的时间加压转移到搅拌(800rpm)的反应混合物中。在有机锂馈料结束时,搅拌速度增加至1130rpm,并且反应器压力从19psig的峰值压力降至14psig的最终压力。在催化剂形成过程期间,温度增加2℃至3℃。最后,将溶解在30g无水乙苯中的3.40g(0.0293摩尔)tmeda与最后50ml无水环己烷等分试样组合,并加压转移到搅拌的反应混合物中。将反应器顶部空间放空至0psig,然后用干燥h2(通过表面下进料管线)加压至45psig。然后将反应器加热至73.2℃,压力增长到63psig。用流经反应器夹套的80℃油进行加热。反应温度达到73℃后,开始苯乙烯单体进料,进料了1058.7g(10.17摩尔)的苯乙烯。经116分钟的时间,相对于11psig的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端)进料,以期将反应温度控制在80℃下。在开始单体进料的10分钟内,反应器温度达到82.8℃。通过关闭调节器的阀门并测定下降5psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计,氢吸收似乎是恒定或接近恒定的。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水环己烷冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml的5重量%h2so4水溶液的洗涤容器中。因此,在洗涤反应器中小心地猝灭反应混合物。然后用300ml无水环己烷漂洗馈料容器和反应器,并且将漂洗溶液转移到洗涤反应器中并与粗猝灭的反应混合物组合。如实施例51在相同的馈料和条件下在按量配给试剂并重现这些条件的最小运行间变化内重复上述过程,不同的是采用10psigh2。尽管h2活性降低了近10%,但实施例51与实施例50相比,氢吸收仍然更快。在转移未猝灭的反应混合物(实施例50和51)期间,获得10ml的各种反应混合物的样品用于分析。样品是微粉色至水白色的-即基本上无色且透光-没有沉降或悬浮的固体。通过轻轻摇动/旋转或以其他方式使混合物与空气接触来猝灭任何颜色。提交样品以进行gpc分析而不猝灭粗反应混合物。排除乙苯但包含二聚物内容物的gpc分析如下:实施例50mn:525,mw:804,mz:1165,pd:1.449,σn=383,nα3=2.075且mw10%高=2048;实施例51mn:506,mw:758,mz:1080,pd:1.425,σn=357,nα3=2.001;并且mw10%高=1844。上述标准后处理提供2613g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)生成1997.5g具有如下包含二聚物的gpcmwd的hmaps分布,mn:519,mw:783,mz:1122,pd:1.452,σn=370,nα3=2.0274;并且mw10%高=1922。第二wfe操作(0.1-0.3mmhg真空,172.5℃,刮擦器速度为全速的60%,以1.0升/小时进料)提供1800.0g具有2.38gpc面积%苯乙烯二聚物含量和如下gpcmwd的hmaps分布,mn:601,mw:836,mz:1142,pd:1.391,σn=326,nα3=1.923;并且mw10%高=1955。实施例52-59实施例52至59证明hmaps方法可以在没有添加的包括乙苯的芳族溶剂的益处情况下运行。这些实施例充分证明hmaps方法非常稳健,因为它在各种工艺条件下可重复地生成几乎相同的hmaps分布。实施例56-59证明,通过使用促进剂tmeda提供的任何工艺益处都可以通过增加氢气压力抵消,从而降低且甚至消除促进剂的使用。这组高收率实施例(聚合物收率为约96%至约97%收率;和二聚物汽提的聚合物的收率为约86%至约87%)生成不对称值在约1.67至2.00的范围内的hmaps分布和不对称值在约1.63至1.82的范围内的二聚物汽提的hmapps分布。实施例58和59被认为是在实施这两个实施例时无意中发现催化剂老化提供更具活性且优选的催化剂的实施例的代表。实施例58和59代表[dmea‾]4li6h2催化剂在80℃下的全刻度单体进料体积在37℃下在干燥氢气(16psigh2)气氛下,将500ml(389.5g)无水环己烷中的200ml馈入充分漂洗的反应器中。向搅拌的溶剂(800rpm,具有上述构型iv的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由2.55g(0.0285摩尔)n,n-二甲基乙醇胺、上述500ml的总共500ml环己烷中的70ml形成的溶液。接下来,将进一步溶解在来自上述500ml的170ml无水环己烷中的21.47ml(0.0429摩尔)2.0m正丁基锂转移到馈料容器中,然后在氢气下经15分钟的时间加压转移到搅拌(800rpm)的反应混合物中。在有机锂馈料结束时,搅拌速度增加至1130rpm,并且反应器压力从19psig的峰值压力减小回到16psig的最终压力。在催化剂形成过程期间,温度增加2℃至3℃。将反应器顶部空间放空至0psig,然后用干燥h2(通过表面下进料管线)加压至45psig。然后将反应器加热至73.2℃,压力增长到63psig。用流经反应器夹套的80℃油进行加热。反应温度达到73℃后,开始苯乙烯单体进料,进料了1061.0g(10.19摩尔)的苯乙烯。经117分钟的时间,相对于15-17psig的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端)进料,将反应温度控制在80℃下。在开始单体进料的10分钟内,反应器温度达到81.7℃。通过关闭调节器的阀门并确定下降5psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计,氢吸收似乎是恒定或接近恒定的。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水环己烷冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml的5重量%无水h2so4和约250g由先前运行的溶剂汽提从刮膜蒸发器回收的乙苯的洗涤容器中。因此,在洗涤反应器中小心地猝灭反应混合物。然后用200ml无水环己烷漂洗馈料容器和反应器,并且将漂洗溶液转移到洗涤反应器中并与粗猝灭的反应混合物组合。如实施例59在相同的馈料和条件下在按量配给试剂的最小运行间变化内重复上述过程,不同的是允许催化剂老化3小时,随后调节h2降至14psig以匹配实施例58的吸收。然而,显而易见的是,除了实施例59具有比实施例58低的不对称性之外,生成的hmaps分布几乎相同;不对称性分别为1.826和1.928。在转移未猝灭的反应混合物(实施例58和59)期间,获得10ml的各种反应混合物的样品用于分析。样品是微粉色至水白色的-即基本上无色且透光-没有沉降或悬浮的固体。通过轻轻摇动/旋转或以其他方式使混合物与空气接触来猝灭任何颜色。提交样品以进行gpc分析而不猝灭粗反应混合物。排除乙苯但包含二聚物内容物的gpc分析如下:实施例58mn:570,mw:890,mz:1276,pd:1.434,σn=427,nα3=1.928且mw10%高=2168;实施例59mn:584,mw:909,mz:1286,pd:1.415,σn=436,nα3=1.826;并且mw10%高=2166。上述标准后处理提供2487g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)生成2094.3g(当调整了洗涤液中自wfe再循环的乙苯的二聚物含量时,2052g)具有如下排除二聚物的gpcmwd的hmaps分布,mn:690,mw:970,mz:1328,pd:1.406,σn=440,nα3=1.889;并且mw10%高=2263。包含反应期间形成的二聚物的总分布在统计学校正时估计如下:mn:612,mw:919.18,mz:1294,pd:1.406,σn=434,nα3=1.8791。第二wfe操作(0.1-0.3mmhg真空,160℃,刮擦器速度为全速的65%,以1.0升/小时进料)提供1825.7g具有0.48gpc面积%苯乙烯二聚物含量和如下gpcmwd的hmaps分布,mn:704,mw:984,mz:1334,pd:1.398,σn=444,nα3=1.815;并且mw10%高=2268。执行第三wfe操作以获得低分子量低聚物,从而确定hmapsps分布微结构。因此,将从第二wfe操作回收的1825g产物分布的125g样品汽提出低聚物(0.13mmhg真空,199.5℃,刮擦器速度为全速的85%,以2.0g/min.进料)。该第三wfe操作生成22g苯乙烯低聚物,其gc分析表明99.97%的链具有期望的“头对尾”微结构,仅痕量(如果有的话)的链具有假定为(fwi-2)的微结构,其中已发生氢化锂消除,生成不饱和链端(参见图16)。真实(fwi-14)材料的强化实验清楚地证明该组合物没有这种微结构。实施例60-67实施例60至67证明hmaps方法可以用从先前实施例获得的干燥(<10ppm水分)再循环的溶剂进行。溶剂的再循环需要组合来自洗涤反应器汽提的环己烷和乙苯和第一wfe操作,共沸蒸馏水,然后在一种气氛下简单蒸馏至约140℃的釜温。使用活化分子筛完成进一步的干燥。由于实施例61和62以及在较低程度上实施例60和67中所采用的工艺条件不太优选,这些实施例在单体进料结束时生成高分子量材料,如通过形成的mwd和在最后的8%至13%进料期间h2吸收减少所证明。在这些运行中,实施例61生成最不优选的结果,形成mw10%高的2629道尔顿的高分子量尾部。对于实施例61,采用较低的催化剂负载量,因此在所得不太优选的相对苯乙烯和催化剂进料速率下,在最后13%的进料期间氢气传质变得低效。实施例60可与实施例58(无老化)和实施例59(老化3小时)相当,不同的是催化剂老化1小时并且溶剂含有再循环的乙苯。最初,催化剂似乎比实施例58或59更具活性;然而,在苯乙烯进料的最后8分钟期间,催化剂活性似乎下降,生成稍高的分子量。对于实施例61,尝试将催化剂负载量降低20%,然而,当在进料的最后13分钟期间氢气吸收减慢时,这产生高分子量尾部。在进一步确立本发明范围的过程中(实施例62-67),发现混合的降低(1000rpm相对标准1130rpm)可以通过以下抵消:i)升高的温度;和/或ii)减少的总单体进料;和/或iii)降低的单体进料速率;和/或iv)增加的氢气压力;v)增加的催化剂老化时间。这些实验的结果呈现在表xii中。实施例64和65被认为是代表性的。实施例64和65代表[dmea‾]4li6h2催化剂在90℃下在从先前实施例回收再循环溶剂的情况下的全刻度单体进料体积在37℃下在干燥氢气(16psigh2)气氛下,将总共320ml(252.65g)回收的无水溶剂[79.4重量%环己烷(ch)、20.6重量%乙苯(eb)]中的220ml馈入充分漂洗的反应器中。向搅拌的溶剂(800rpm,具有上述构型iv的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入预先由2.55g(0.0285摩尔)n,n-二甲基乙醇胺和进一步与50ml来自上面的再循环溶剂组合的20ml环己烷形成的溶剂。接下来,将溶解在120ml无水环己烷中的21.47ml(0.0429摩尔)2.0m正丁基锂转移到馈料容器中,并将其与50ml来自上面的再循环溶剂(很少(如果有的话)反混合)组合。然后在氢气下经15分钟的时间将该未混合的溶液加压转移到搅拌(800rpm)的反应混合物中。在有机锂馈料结束时,搅拌速度增加至1130rpm,并且反应器压力从19psig的峰值压力减小回到16psig的最终压力。在催化剂形成过程期间,温度增加2℃至3℃。将反应器顶部空间放空至0psig,然后用干燥h2(通过表面下进料管线)加压至45psig。然后将反应器加热至72℃,压力增长到63psig并且进一步加压到72psig。用流经反应器夹套的80℃油进行加热-将反应器保持在72℃和72psig下,持续60分钟,然后放空到17psig。然后将反应器加热至82℃,通过根据需要放空使压力维持在17psig。用流经反应器夹套的90℃油进行加热-将反应器保持在82℃和17psig下,持续75分钟,此时开始苯乙烯单体进料,进料1015.0g(9.75摩尔)苯乙烯。经119分钟的时间,相对于13-15psig的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端)进料,将反应温度控制在80℃下。在开始单体进料的10分钟内,反应器温度达到81.7℃。通过关闭调节器的阀门并确定下降5psig所需的时间来定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计,氢吸收似乎是恒定或接近恒定的。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水环己烷冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml的溶解在300gh2o中的5.13g乙酸以及300预先由先前运行的初始溶剂汽提回收的回收溶剂的洗涤容器中。因此,在洗涤反应器中小心地猝灭反应混合物。然后用200ml的再循环溶剂漂洗馈料容器和反应器,并且将漂洗溶液转移到洗涤反应器中并与粗猝灭的反应混合物组合。如实施例65在相同的馈料和条件下在按量配给试剂的最小运行间变化内的重复上述过程,不同的是经121分钟进料1035.8g苯乙烯,允许催化剂在72℃下老化34小时并且在82℃下老化26分钟。很明显,生成的hmaps分布与gpc分析的实验误差内几乎相同。在转移未猝灭的反应混合物(实施例64和65)期间,获得10ml的各种反应混合物的样品用于分析。样品是微粉色至水白色的-即基本上无色且透光-没有沉降或悬浮的固体。通过轻轻摇动/旋转或以其他方式使混合物与空气接触来猝灭任何颜色。提交样品以进行gpc分析而不猝灭粗反应混合物。排除乙苯但包含二聚物内容物的gpc分析如下:实施例64mn:550,mw:835,mz:1173,pd:1.405,σn=396,nα3=1.832且mw10%高=1982;实施例65mn:558,mw:848,mz:1188,pd:1.401,σn=402,nα3=1.805;并且mw10%高=2003。上述标准后处理提供2487g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)产生1947g具有如下包含二聚物的gpcmwd的hmaps分布,mn:562,mw:848,mz:1186,pd:1.509,σn=401,nα3=1.811;并且mw10%高=2002。第二wfe操作(0.1-0.3mmhg真空,160℃,刮擦器速度为全速的65%,以1.0升/小时进料)提供1716g具有0.13gpc面积%苯乙烯二聚物含量和如下gpcmwd的hmaps分布,mn:695,mw:935,mz:12284,pd:1.345,σn=408,nα3=1.681;并且mw10%高=2064。执行第三wfe操作以获得低分子量低聚物,从而确定hmapsps分布微结构。因此,将从第二wfe操作回收的1825g产物分布的125g样品汽提出低聚物(0.13mmhg真空,199.5℃,刮擦器速度为全速的85%,以2.0g/min进料)。该第三wfe操作生成22g苯乙烯低聚物,其gc分析表明99.93%的链具有期望的“头对尾”微结构,仅痕量(如果有的话)的链具有假定为(fwi-2)的微结构,其中已发生氢化锂消除,生成不饱和链端(参见图17)。实施例68-74实施例68至74证明hmaps方法可以用从先前实施例获得的100%干燥的(<10ppm水分)再循环的溶剂进行。如上述,溶剂的再循环需要组合来自洗涤反应器汽提的环己烷和乙苯和第一wfe操作,共沸蒸馏水,然后在一种气氛下简单蒸馏至约140℃的釜温。使用活化分子筛完成进一步的干燥。如上述一样,由于实施例68和72所采用的工艺条件不太优选,对于所采用的反应器几何形状以<1000rpm进行混合;并且在较小程度上实施例67和70在单体进料结束时生成高分子量材料,如通过所形成的mwd以及在最后8%至13%进料期间h2吸收降低所证明。重要的是要注意,实施例68和69与上述实施例66和67几乎相同,不同的是在馈料之前用以进一步稀释2.0m正丁基锂的溶剂的变化。在实施例66和实施例67中,使用新鲜的无水环己烷以初步溶解有机锂试剂。而在实施例68和69中,采用由93%环己烷和7%乙苯组成的再循环溶剂。这种馈料方案令人惊讶地形成了一种活性高得多的催化剂,如通过在甚至更低的氢气压力(分别地,17psig至19psig相对13psig至14psig)下略低的分子量分布(mn=550和mn=558[分别地,实施例64和65]对比mn=532和mn=533[分别地,实施例68和69)所证明。实施例70-73证明,通过降低搅拌器的rpm并因此降低聚合期间的混合量,可以缩减通过首先用一定量的乙苯稀释形成的催化剂的活性(在该方法中没有使用任何促进剂)。然而,这是不太优选的,因为倾向于在苯乙烯进料结束时形成高分子材料,这可以通过增加rpm和/或氢气压力和/或降低进料的苯乙烯单体的总量来抵消。实施例72和73证明,与在略微降低的rpm下形成催化剂相比,存在更多的乙苯具有较小的影响。因此,催化剂通常以800rpm形成,这足以在用本发明的反应器几何形状形成催化剂期间实现氢向凝相的有效传质。对于实施例73,将催化剂组分在500rpm混合下组合,其远小于800但显然足以令人惊讶地形成甚至具活性的催化剂。在实施例74中,使用200rpm和2psig氢气来形成催化剂。该催化剂最初活性很大,并且必须在9psig至11psig下且甚至在氢气活性h2吸收太大的情况下运行。令人惊讶的是,这种催化剂虽然在开始时活性很大,但活性越来越低,生成低分子量链高的非常不对称分布,并且mw10%高为3323道尔顿。因此,当在远高于低温条件的温度下形成催化剂时,期望减少由[dmea‾]li和正丁基锂形成的络合物。尽管我们希望不受理论束缚,但认为通过控制混合和氢气传质来控制还原速率有助于预处理催化剂以在烃溶液中形成最具活性形式的催化剂聚集体。因此,实施例72和73被认为是这组实施例(68-74)的代表,并在下面更详细地描述。实施例72和73代表[dmea‾]4li6h2催化剂在90℃下在用100%从先前实施例回收的再循环溶剂的情况下的完全和90%刻度单体进料体积在37℃下在干燥氢气(16psigh2)气氛下,将总共500ml(392g)再循环的无水溶剂中的220ml[85.0重量%环己烷(ch)、15.0重量%乙苯(eb)]馈入充分漂洗的反应器中。向搅拌的溶剂(800rpm,具有上述构型iv的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入由2.54g(0.0285摩尔)n,n-二甲基乙醇胺和20ml来自上面的再循环溶剂形成的充分混合的溶液,当在所述馈料容器中组合时在少许混合(如果有的话)下向其中添加另外50ml再循环溶剂等分试样。接下来,将进一步溶解在120ml再循环溶剂中的21.47ml(0.0429摩尔)2.0m正丁基锂转移到馈料容器中,并将其与50ml来自上面的再循环溶剂进一步组合。然后在氢气下经15分钟的时间将该未混合的溶液加压转移到搅拌(800rpm)的反应混合物中。在有机锂馈料结束时,搅拌速度增加至1130rpm,并且反应器压力从19psig的峰值压力减小回到16psig的最终压力。在催化剂形成过程期间,温度增加2℃至3℃。将反应器顶部空间放空至0psig,然后用干燥h2(通过表面下进料管线)加压至45psig。然后将反应器加热至72℃,压力增长到63psig并且进一步加压到72psig。用流经反应器夹套的80℃油进行加热-将反应器保持在72℃和72psig下,持续150分钟,然后放空到17psig。然后用流经反应器夹套的90℃油将反应器加热至80℃,其中压力增加到20psig。在150分钟催化剂老化结束时,将rpm调节至960时并开始苯乙烯单体进料,进料1000.0g(9.60摩尔)的苯乙烯。经122分钟的时间,相对于15-17psig的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端)进料,控制反应温度在91℃下。在开始单体进料的10分钟内,反应器温度达到90.5℃。通过关闭调节器的阀门并测定下降5psig所需的时间定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计,氢吸收似乎是恒定或接近恒定的。在运行结束时,将rpm调节至975并且氢气压力增加至18psig。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水环己烷冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入300ml的溶解在300gh2o水中的5.13g乙酸以及300预先由先前运行的初始溶剂汽提回收的回收溶剂的洗涤容器中。因此,在洗涤反应器中小心地猝灭反应混合物。然后用200ml的再循环溶剂漂洗馈料容器和反应器,并且将漂洗溶液转移到洗涤反应器中并与粗猝灭的反应混合物组合。如实施例73在相同的馈料和条件下在按量配给试剂的最小运行间变化内重复上述过程,不同的是相对于14psig的氢气压力,经111分钟进料911.3g的苯乙烯,将催化剂组分在500rpm混合而不是800rpm混合下组合。进料量的减少排除在进料结束时对增加rpm或氢气压力的需要。很清楚,除了实施例72在进料结束时生成少量高分子量组合物外,生成的hmaps分布非常相似。在转移未猝灭的反应混合物(实施例72和73)期间,获得10ml各种反应混合物的样品用于分析。样品是微粉色至水白色的-即基本上无色且透光-没有沉降或悬浮的固体。通过轻轻摇动/旋转或以其他方式使混合物与空气接触来猝灭任何颜色。提交样品以进行gpc分析而不猝灭粗反应混合物。排除乙苯但包含二聚物内容物的gpc分析如下:实施例72mn:485,mw:747,mz:1133,pd:1.517,σn=356,nα3=2.462且mw10%高=1965;实施例73mn:480,mw:710,mz:1009,pd:1.421,σn=332,nα3=2.025;且mw10%高=1732。上述标准后处理提供2270g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)生成1786.5ghmaps,其在第二wfe操作(0.1-0.3mmhg真空,160℃,刮擦器速度为全速的65%,以1.0升/小时进料)进一步汽提(没有分析),提供1574g具有0.13gpc面积%苯乙烯二聚物含量和如下gpcmwd的hmaps分布,mn:591,mw:817,mz:1149,pd:1.382,σn=365,nα3=2.285;并且mw10%高=1998。完成实施例42至74(总共32次运行)后,用1升不含有机物用氮气吹扫的循环溶剂冲洗反应器,然后打开检查。发现反应器通常非常干净,在壁和表面上没有聚合物且仅有痕量水平的固体,该水平标志着用于形成催化剂的溶剂的体积。用湿布将反应器擦拭干净,然后用由甲醇润湿的布擦拭。将单体0.02”内径进料尖端用长度相等但内径为≈0.45”的进料尖端替换。将反应器密封,在氮气流下干燥,同时用100℃油加热夹套。实施例75-79在所有这些实施例(75至79)中,催化剂在500rpm混合下在18-21psig氢气压力下形成,在72℃和72psig氢气压力下进一步老化200分钟至240分钟的时间。实施例75至77利用0.045”内径单体进料管线尖端、80℃和1130rpm混合、不同的苯乙烯进料速率和不同的再循环溶剂馈料和组合物。在这三次运行中,优选实施例77的hmaps方法。实施例78利用0.045”内径苯乙烯进料尖端和新鲜的无水甲基环己烷(mch)溶剂,因此在该运行期间,没有用乙苯初始稀释有机锂试剂。完成实施例75-78后,再次冲洗反应器并用氮气吹扫有机物并打开检查。发现反应器是清洁的,并且在催化剂形成期间标记反应器体积的线处没有任何固体。据推测,在催化剂形成期间采用的较低rpm也可以带来催化剂形成反应混合物较少飞溅的益处,从而减少催化剂组分由于溶剂从暴露并加热的反应器表面闪蒸出而引起的沉积。完成实施例75-78后,除去不太优选的0.045”内径苯乙烯进料尖端,并用优选的0.02”苯乙烯进料尖端代替,然后准备用于在该系列的最终运行(实施例79)。利用在mch和乙苯中形成的催化剂的实施例79在下面更详细地描述。实施例79代表[dmea‾]4li6h2催化剂在90℃下在用混合的乙苯和甲基环己烷溶剂的情况下的全刻度单体进料体积在37.6℃下在干燥氢气(18psigh2)气氛下,将总共320ml(246.4g)无水甲基环己烷(mch)溶剂中的220馈入充分漂洗的反应器中。向搅拌的溶剂(500rpm,具有上述构型iv的四节距叶片涡轮机)中通过正氮气压力经馈料容器馈入由2.55g(0.0286摩尔)n,n-二甲基乙醇胺和20ml的乙苯形成的充分混合的溶液,当在所述馈料容器中组合时在少许混合(如果有的话)下向其中添加50mlmch等分试样。接下来,将进一步溶解在160ml无水乙苯中的21.52ml(0.0430摩尔)2.0m正丁基锂转移到馈料容器中,并将其与50mlmch在少许(如果有的话)反混合下进一步组合。然后在氢气下经20.75分钟的时间将该未混合的溶液加压转移到搅拌(525rpm)的反应混合物中。在有机锂馈料结束时,搅拌速度增加至1050rpm,并且在38.8℃下反应器压力从21psig的峰值压力减小回到20psig的最终压力。然后反应器顶部空间用干燥h2(通过表面下进料管线)加压到48psig。然后将反应器加热至73.2℃,压力增长到59psig,并进一步加压至75psig(自从完成有机锂向反应器中的进料以来已过去60分钟)。用流经反应器夹套的80℃油进行加热-将反应器保持在73.9℃和75psig下,持续205分钟,然后放空到15psig。因此自开始将在乙苯中的有机锂试剂溶液馈料到反应器以来已过去约4小时。此时,将混合物调节至1130rpm并开始苯乙烯单体进料,进料1023.9g(9.83摩尔)的苯乙烯。经113分钟的时间,相对于11-15psig的氢气顶部压力,苯乙烯经表面下进料管线(0.02”内径尖端)进料,控制反应温度在82.5℃下。在开始单体进料的10分钟内,反应器温度达到83.6℃。通过关闭调节器的阀门并测定下降5psig所需的时间定期监测氢吸收。因此,记录压力下降(-1)1psig所需的时间(秒)。当针对估计的反应器顶部空间调节该值时,以摩尔h2/摩尔苯乙烯进料计,氢吸收似乎是恒定或接近恒定的。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱(碱性氧化铝),用50ml无水mch冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正h2压力下转移到预先加热(n2气氛)并预先馈入400ml的溶解在380gh2o水中的6g乙酸和14gh2so4以及300ml新鲜mch的洗涤容器中。因此,在洗涤反应器中小心地猝灭反应混合物。然后用200ml的再循环溶剂漂洗馈料容器和反应器,并且将漂洗溶液转移到洗涤反应器中并与粗猝灭的反应混合物组合。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品用于分析。样品是微粉色至水白色的-即基本上无色且透光-没有沉降或悬浮的固体。通过轻轻摇动/旋转或以其他方式使混合物与空气接触来猝灭任何颜色。提交样品以进行gpc分析而不猝灭粗反应混合物。排除乙苯但包含二聚物内容物的gpc分析如下:mn:511,mw:767,mz:1096,pd:1.429,σn=362,nα3=2.020,并且mw10%高=1871。上述标准后处理提供1000g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器速度为全速的60%,以1.0升/小时进料)生成961g具有如下gpc分析的hmaps分布::mn:542,mw:804,mz:1142,pd:1.483,σn=377,nα3=2.009,并且mw10%高=1955。第二wfe操作(0.1-0.3mmhg真空,160℃,刮擦器速度为全速的65%,以1.0升/小时进料)提供871g具有1.3gpc面积%苯乙烯二聚物含量和如下gpcmwd的hmaps分布,mn:627,mw:855,mz:1163,pd:1.364,σn=378,nα3=1.999;并且mw10%高=1995。由实施例42-79制备的hmaps分布根据pct公告wo2010/127091(us8,802,787b2)的工艺技术溴化以形成溴化的阴离子链转移乙烯基芳族聚合物(br-actvap)。下面列出这些溴化聚合物的物理特性的平均值和标准偏差,以及通过组合然后汽提实施例14-15的产物分布形成的br-actvap的特性,该二聚物汽提的组合物具有mw=731和pdn=1.38。这些组合物在高抗冲聚苯乙烯(hips)中作为聚合物阻燃剂进一步测试,并且发现提供阻燃(ul94v0,在1/8”和1/16”下)hips制剂,其具有优异的整体特性,包括颜色(yi)、悬臂梁式冲击、热变形温度和vicat软化温度比较例在下面给出并在上面图12、13和14中说明的比较例80-83中,在氮气氛下使用现有技术的阴离子链转移聚合方法,其中链转移剂是乙苯并且使用钾基催化剂,据证明这些现有技术极大地遭受不期望的竞争聚合步骤的问题,这些步骤导致形成具有不同且不太期望的微结构的复杂聚合物链分布(即,图1和2中提供的反应途径的步骤)。在实施例25-27、39和40中,我们证明loxkh、sash和hash、钾和钾与钠基催化剂体系、本发明的氢介导的阴离子链转移工艺技术如(无氢)现有技术生成复杂的多聚合物链长分布。这种基于钾和钠的类盐氢化物工艺产物分布组合物之间的差异主要在于期望的“头对尾”和不期望的“尾对头对尾”及断裂微结构的不同相对比例。如实施例1-24、28、29和42-79中所述,不可预测且有益的是,采用loxlih和/或loxmgh2聚集体作为催化剂的本发明的工艺技术提供了大大改善的整体聚合物微结构。也就是说,loxlih和loxmgh2催化剂生成几乎仅由最期望的“头对尾”微结构的低聚物链组成的聚苯乙烯分布。令人惊讶的是,本发明的这些试剂和催化剂出于所有意图和目的几乎消除了所有不期望的竞争途径,并且仅提供在已经证明可用于形成聚合物产物如聚合物溴化阻燃剂的分子量分布范围内的最期望的聚合物微结构(即,上述聚合物结构1)的链转移分布。尽管我们希望不受理论束缚,但在阴离子链转移聚合条件下,当采用除loxlih和/或loxmgh2之外时,显著的竞争性副反应途径产生具有不同且不期望的微结构的异构低聚链的额外分布(参见图1)。图1的异构化途径提供需要断裂/聚合的额外竞争途径。这些断裂/聚合过程导致:(a)具有增加一个甲基(-ch3)碳的低聚物链结构(增加14道尔顿);和/或(b)具有减少一个甲基(-ch3)碳的低聚物链结构(减少14道尔顿,参见图2)。这种竞争性聚合反应途径是不期望的,因为它们通常导致形成聚合物链主链(即,聚合物微结构)中的季碳原子。这种季碳原子使聚合物分布与芳族亲电取代催化剂不太相容,这些芳族亲电取代催化剂包括布朗斯酸(例如,磺化)或路易斯酸(例如,卤化)。比较例80自wo2010065468a8的比较例46、epo741147的乙苯链转移组合物分离低分子量低聚物将自wo2010065468a8的实施例46保留的150ml水洗产物混合物样品在kugelrohr蒸馏装置中在减压下小心地浓缩。在将环己烷溶剂和乙苯链转移剂从聚合物中蒸馏至kugelrohr烘箱中150℃和灯泡管中1.0mmhg真空的最终条件。取出带有溶剂的接收器并用新的接收器替换,继续蒸馏直至烘箱中的最终温度为220℃并且在灯管中<0.1mmhg真空,继续蒸馏直到没有迹象表明可以觉察到在接收器中有凝结物。将接收器的内容物溶解在二氯甲烷中并通过气相色谱分析。gc分析表明,93%的链具有期望的“头对尾”微结构,3.4%的链具有不期望的“尾对头对尾”季碳键合,并且3.2%的链具有断裂的(fwi-14)微结构(参见图12)。比较例81使用低聚物微结构分析自wo2008154453的actvap组合物中分离低分子量低聚物actvap组合物由苯乙烯制备,其中将乙苯而不是甲苯用作链转移剂。阴离子链转移方法在wo2008154453中描述的12升玻璃聚合反应器中以与该申请的实施例8类似的方式进行。因此,将1905g的苯乙烯(18.29摩尔)进料(110分钟,70℃反应温度)到由在4517.07g(42.55摩尔)中由13.74g(0.1224摩尔)叔丁醇钾、71.51g(0.6154摩尔)tmeda和63.12(0.1262摩尔)的2m在环己烷中的正丁基锂形成的反应混合物中。在后处理并汽提出乙苯(wfe195℃,60mmhg)后,回收2765.7g具有如下gpcmwd的actvap:mn:266,mw:301,mz:357,pd:1.133。第二wfe操作(195℃,25mmhg)产生2070g具有以下gpcmwd的actvap组合物:mn:297,mw:334,mz:385,pd:1.126。对来自第一的2070g中的1106g进行第三wfe操作(135℃,0.5mmhg)产生909.25g基本上不含该方法的主要反应产物的组合物,其为乙苯与苯乙烯的单加合物(结构上与头对尾苯乙烯二聚物相同)。第四wfe操作(199.5℃,0.12mmhg)产生449.02g的馏出物。gc分析表明,91%的链具有期望的“头对尾”微结构,8.9%的链具有不期望的“尾对头对尾”季碳键合,且0.22%的链具有断裂的(fwi-14)微结构(参见图13)。比较例82与实施例40相同的方法,不同之处在于在氮气下-没有氢气,使用低聚物微结构分析在干燥氮气氛下将356g(3.35摩尔)无水乙苯中的256g馈入反应器中,然后加热到65℃。向搅拌的溶剂(1000rpm,双节距叶片叶轮,叶片放置构造i)中经馈料容器馈入预先由4.57g(0.0407摩尔)叔丁醇钾、44g(0.41摩尔)乙苯和20.83g(0.179摩尔)的tmeda形成的溶液。将馈料容器和反应器的转移管线用上述356g无水乙苯的50g份冲洗。接下来,将20.34ml(0.0407摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述的50g乙苯等分试样。将搅拌维持在1045rpm下,同时经183分钟的时间,相对于氮气顶部压力,将800g(7.68摩尔)苯乙烯进料经过表面下进料管线(0.01”内径尖端,5.2ft/s),将温度控制在70℃下。在进料期间,反应器压力从0psig增加到4psig,然后放空回到0psig,因此对于剩余的进料,将反应器排放到矿物油鼓泡器中。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱,用50ml无水环己烷冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正n2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水以及500ml环己烷的洗涤容器中。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品用于分析。样品为深黑红色(黑樱桃色),并且不透光,透光是先前观察到的所有其他actvap和actsp过程所特有的。通过添加一滴甲醇猝灭样品,该甲醇立即猝灭深红色,而没有形成气体。粗猝灭反应混合物的gpc分析如下:mn:567,mw:908,mz:1331,pd:1.601,σn=440,nα3=2.048。上述标准后处理和溶剂带提供1240.2g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器全速的60%,以1.0升/小时进料)生成943.1g具有如下gpcmwd的actvap分布,mn:580,mw:904,mz:1286,pd:1.559,σn=433,nα3=1.868。第二wfe操作(0.1-0.3mmhg真空,172.5℃,刮擦器全速的60%,以1.0升/小时进料)提供849.4具有0.70gpc面积%苯乙烯二聚物含量和如下gpcmwd的actvap分布,mn:707,mw:976,mz:1306,pd:1.380,σn=436,nα3=1.741。因此,除了采用的气氛外,该比较例的条件与实施例40的条件是相同的。显然,实施例40的氢介导大大改善了链转移效率,并提供mn、mw和mz值低得多的mwd。执行第三wfe操作以获得低分子量低聚物,以确定actvap分布微结构。因此,将从第二wfe操作回收的849.4g产物分布的131.2g样品汽提出低聚物(<0.1mmhg真空,199.5℃,刮擦器速度为全速的85%,以2.0g/min进料)。该第三wfe操作生成19.07g具有如下mwd的苯乙烯低聚物混合物,mn:318,mw:338,mz:357,pd:1.061。gc分析表明,93%的链具有期望的“头对尾”微结构,5.0%的链具有不期望的“尾对头对尾”季碳键合,并且2.3%的链具有断裂的(fwi-14)微结构(参见图14)。比较例83与实施例40相同的方法,不同之处在于在氮气下和比较例43无氢气但使用正丙基苯使用低聚物微结构分析在干燥氮气氛下将381g(3.18摩尔)无水正丙基苯中的281g馈入反应器中,然后加热到65℃。向搅拌的溶剂(1130rpm,双节距叶片叶轮,叶片放置构造i)中经馈料容器馈入预先由4.61g(0.0411摩尔)叔丁醇钾、50g(0.42摩尔)正丙基苯和26.34g(0.179摩尔)的tmeda形成的溶液。将反应器的馈料容器和转移管线用上述381g无水正丙基苯的50g份冲洗。接下来,将20.32ml(0.0407摩尔)2.0m正丁基锂经馈料容器转移到反应器中,然后加入来自上述的50g正丙基苯等分试样。将搅拌维持在1131rpm下,同时经183分钟的时间,相对于氮气顶部压力,804g(7.72摩尔)苯乙烯经表面下进料管线(0.01”内径尖端,5.2ft/s)进料,控制温度在70℃下。在苯乙烯单体进料期间,使反应器压力从0psig增加到9psig。在苯乙烯进料结束时,将反应器的单体进料管线,包括氧化铝柱,用50ml无水环己烷冲洗。当没有观察到进一步的反应热时,这通常通过卷绕盘管上的自动控制阀的永久关闭所预示,认为对反应器的苯乙烯进料和冲洗完成。将未猝灭的聚合反应混合物在正n2压力下转移到预先加热(n2气氛)并预先馈入300ml脱氧水以及500ml环己烷的洗涤容器中。在转移未猝灭的反应混合物期间,获得10ml反应混合物样品用于分析。样品为深黑红色(黑樱桃色),并且不透光,透光是先前观察到的所有其他actvap和actsp过程所特有的。通过添加一滴甲醇猝灭样品,该甲醇立即猝灭深红色,而没有形成气体。粗猝灭反应混合物的gpc分析如下:mn:668,mw:1013,mz:1354,pd:1.517,σn=480,nα3=1.413。上述标准后处理和汽提生成1908.2.2g的溶液。刮膜蒸发(wfe,2”玻璃popestill,在50.0mmhg真空下操作,140℃,刮擦器全速的60%,以1.0升/小时进料)。该第一wfe操作生成939.88g具有如下gpcmwd的actvap分布,mn:690,mw:1017,mz:1336,pd:1.475,σn=475,nα3=1.992。第二wfe操作(0.1-0.3mmhg真空,172.5℃,刮擦器全速的60%,以1.0升/小时进料)提供866.04具有0.70gpc面积%苯乙烯二聚物含量和如下gpcmwd的actvap分布,mn:785,mw:1066,mz:1353,pd:1.358,σn=470,nα3=1.245。因此,除了采用的链转移剂之外,该比较例的条件和比较例43的条件是相同的,因此组合物在其mwd方面几乎相同。执行第三wfe操作以获得低分子量低聚物,以确定actvap分布微结构。因此,将从第二wfe操作回收的866.04g产物分布的161.4g样品汽提出低聚物(<0.1mmhg真空,199.5℃,刮擦器速度为全速的85%,以2.0g/min进料)。该第三wfe操作生成16.33gactvap低聚物混合物。gc分析表明99.08%的链具有期望的“头对尾”微结构(参见图9)。因此,从这三个比较例(41-43)中可以清楚地看出,依赖乙苯作为链转移剂的现有技术组合物在其形成中遭受导致形成具有不期望的微结构的聚合物链的额外分布的两种链异构化反应。比较例和本发明的loxkh、sash和hash实施例中呈现的结果表明,钾和钠促进了图1和图2中所示的这种不期望的途径。相当令人惊讶且有益的是,本发明的loxlih和loxmgh2实施例清楚地表明,当在同样是本发明的氢介导的类盐氢化物引发的聚合方法中采用这些新型催化剂时,图1和2的反应途径被大大抑制并且甚至出于所有意图和目的而被消除。重要的是要注意,当聚合物链引发物质是α-甲基苄基阴离子arc(r)h‾(其中r=ch3)时,仅产生不期望的高水平-大于3%的聚合物链-季碳,而不管是由乙苯形成还是由苯乙烯形成。对于其中r=h的阴离子arc(r)h‾(即,由甲基苯如甲苯形成的引发剂),无论多么频发地发生类似于图1的路径,在结构上都不可能形成季碳键合。如比较例44所证明,对于阴离子arc(r)h‾,当r是大于ch3的烷基(例如,如实施例40中,ch2ch3)并且同样诱导性地比ch3更好的电子供体时,图1和2的途径显著减少,因此可以产生具有少于3%甚至少于1%的具有季碳键合的链的组合物。通过检查以下结构,应该清楚地知道这一点:活跃的苯乙烯三聚物活跃的苯乙烯五聚物。如现在可以根据本发明的发现来解释,应该进一步理解,图1的异构化反应-回咬(backbiting)途径-基本上仅对于苯乙烯的活跃的三聚物发生。本领域普通技术人员将认识到涉及活跃的苯乙烯四聚物、五聚物、六聚物等的后回咬途径将迫使苯基基团或聚(苯乙烯基)链进入不利的位置,因此抑制了回咬。从描绘上述活跃五聚物的化学结构中可以清楚地看出这一点。因此,由回咬或分子内质子转移引起的不期望的链异构化反应的程度由立体电子效应决定。过渡态或活化的络合物包含6个原子(5个碳和1个质子),并且当要转移的质子的α基团较大且电子供体较强时,其被抑制。令人惊讶的是,与阴离子相关的阳离子的性质对质子转移反应具有剧烈的影响。因此,与其他碱金属和碱土金属相比,活跃的三聚物的分子内质子转移被镁大大抑制并且被锂基本上消除。因此,钠、钾和所有其他第1族和第2族金属促进不期望的分子内质子转移,并且这种回咬反应生成具有不太期望的聚合物微结构的组合物。当由乙苯以及苯乙烯或由单独的苯乙烯形成组合物时,这尤其成问题。当使用基于锂的链转移催化剂(例如,由乙苯、丁基锂和tmeda形成的催化剂,来自epo741147的上述实施例d的表i)时,乙苯是无效的链转移剂。然而,与本发明的新型氢介导的类盐氢化物引发的聚合方法组合使用的新型单金属锂和新型双金属锂和镁催化剂首次提供如下阴离子链转移聚苯乙烯分布:不含额外的有机链转移剂且具有非常高-大于97%,甚至大于99.2%-期望的“头对尾”聚合物微结构并且基本上没有并入除氢化物离子外的其他引发物质-这种微结构完整性对于经由聚苯乙烯组合物的芳族亲电取代反应进行进一步衍生化是必需的。分析方法对于低分子量(mw<1600道尔顿),以mw、mn、mz、pd和mw10%高值而言的分子量分布通过gpc使用配备有uv检测器、自动进样器、泵和温控柱隔室的viscotektda模块化系统获得。使用的柱是agilentoligopore柱,300mm×7.5mm,部件号1113-6520。使用的溶剂是hplc级的四氢呋喃。使用的测试程序需要将约0.06-0.1g的样品溶解在10ml的thf中。过滤该溶液的等分试样并在柱上注入200μl。基于分离的1,3-二苯基丁烷(二聚物)和1,3,5-三苯基己烷(三聚物)加合物,并且分离模式是尺寸排阻,根据它们的洗脱顺序鉴定峰为1,3-二苯基丁烷、13,5-三苯基己烷、1,3,5,7-四苯基辛烷(四聚物)、1,3,5,7,9-五苯基癸烷(五聚物)等。然后将低聚物材料的各个峰指定为理论分子量值。使用这些理论值及其相应的保留时间构建校准曲线。基于该校准,计算并报道总体分布数据。计算由viscotekomnisec,4.2.0.237版凝胶渗透色谱(gpc)数据收集和处理系统进行。对于较高分子量(mw>1600道尔顿),以mw、mn、mz和pd而言的分子量分布通过gpc使用配备有uv检测器、自动进样器、泵和温控柱隔室的viscotektda模块化系统获得。以下三个agilenttechnologies柱串联使用以执行分离:(1)oligopore柱,300mm×7.5mm,部件号1113-6520,(1)混床e,300mm×7.5mm,部件号1110-6300,和(1)混床d,300mm×7.5mm,部件号1110-6504。使用的溶剂是hplc级的四氢呋喃。使用的测试程序需要将约0.06-0.1g的样品溶解在10ml的thf中。过滤该溶液的等分试样并在柱上注入200μl。基于分离的1,3-二苯基丁烷(二聚物)和1,3,5-三苯基己烷(三聚物)加合物,并且分离模式是尺寸排阻,根据它们的洗脱顺序鉴定峰为1,3-二苯基丁烷、13,5-三苯基己烷、1,3,5,7-四苯基辛烷(四聚物)、1,3,5,7,9-五苯基癸烷(五聚物)等。然后将低聚物材料的各个峰指定为理论分子量值。使用这些理论值及其相应的保留时间以及已知分子量的聚苯乙烯参考标准物的保留时间构建校准曲线。基于该校准,计算并报道总体分布数据。如上述,计算由viscotekomnisec,4.2.0.237版凝胶渗透色谱(gpc)数据收集和处理系统进行。用于分析低分子量苯乙烯低聚物(二聚物至六聚物)的气相色谱法和条件如下。使用配备有agilenttechnologiesdb-530计,0.25mm内径,0.25μm柱的hewlettpackardhp6850气相色谱仪分析从产物树脂的刮膜蒸馏和/或kugelrohr蒸馏获得的苯乙烯低聚物混合物。将低聚物样品制备成2.5重量%的二氯甲烷溶液并手动注入(注射温度为270℃),使用具有氦载气的温度程序分离并使用火焰离子化检测器测量响应。温度程序如下:a)100℃初始温度保持2分钟,其中载气流速1.5ml/min;b)以8℃/min的速度升温至300℃,其中载气流速为2.0ml/min;c)在300℃下保持10.0分钟,其中载气流速为2.0ml/min;d)以3.0℃/min按程序升温至320℃,其中载气流速为2.0ml/min;以及e)在320℃下保持15.0分钟,其中载气流速为2.0ml/min。收集数据并使用atlas8.2.3色谱数据系统分析。基于分离的标准物或标准物混合物对基于并入低聚物微结构中的苯乙烯单体单元的数量分组(即,二聚物与三聚物分开,与四聚物分开,与五聚物分开,与六聚物分开)并且基于该组的总面积计数归一化的低聚物进行微结构分配。另外,通过质谱法分析低聚物混合物,以进一步确认由上述图1和2的竞争性不期望的断裂聚合过程形成的低聚物结构的分配(即,fwi=[i(104)+2–14]道尔顿的离散低聚物以及fwi=[i(104)+2+14]的离散低聚物,其中i是以下数字:1)苯乙烯单体单元;或2)乙苯苯乙烯链转移聚合产物的芳环数;并入所关注的离散低聚物链中)。如图3-17中所证明,从该微结构gc方法的应用中清楚且明确地看出,组成相关但又完全不同的现有技术对于前六种离散低聚物结构提供具有截然不同和不期望量的低聚微结构的组合物。此外,认为这种分析二聚物至六聚物产物混合物,特别是三聚物和四聚物低聚物的技术可预测整个聚合物分布的微结构完整性或纯度。因此,充分地认为整个分布的微结构纯度可以由该低聚物分析确立。因此,优选的loxshps和hmaps组合物通过低聚物测试容易地与现有技术组合物区分开,无论gc分析是对前5种(二聚物至六聚物)低聚物进行,还是仅对三聚物和四聚物进行。这种微结构纯度被认为是优于现有技术的优越优点,并且是形成通过聚合物的oecd定义视为聚合的并且基本上由(如果不是唯一的话)苯乙烯单体组成的聚苯乙烯的一项进步。确定本发明的催化剂和试剂组合物的经验式如上所述,在本领域中已经很好地确立了烷基锂化合物作为引发剂以及混合的有机金属作为引发剂用于苯乙烯和共轭二烯的活跃阴离子聚合反应的缔合度所带来的复杂性(在这方面,参见hsiehh.l.和quirk,r.p.anionicpolymerizationprinciplesandpracticalapplications,marceldekker,1996,newyork,第135-132页,特别是表6.2,138页)。正丁基锂的缔合度通常为6,其中叔丁基锂和仲丁基锂的缔合度在烃溶剂中通常为4。因此,缔合的烷基锂化合物的聚集体中仅六分之一的正丁基锂试剂引发活跃聚合,对于仲丁基锂和叔丁基锂两者而言,仅四分之一引发活跃的聚合。本发明的一个概念是,类似地,每个催化剂组合物聚集体中仅一个氢化物将引发阴离子聚合。因此,形成的聚合物的数量与理论上可以在活跃阴离子聚合下形成的聚集体数量的反比将为本发明的平均构成催化剂组合物提供证据。如上所述,仅两种已知的烃可溶性氢化锂试剂以聚集体形式存在:通过光降解形成的超级聚集体具有分子式[(t-buoli)16(lih)17];和stash的烃可溶性lih络合物具有分子式[(dipnpph2)4li8h4](dip,2,6-ipr2c6h3)。在两种情况下,分子式通过分离和x射线晶体学确定。不必分离本发明的催化剂组合物,从而可以进行x射线晶体学或者说燃烧分析或其他现代化学分析方法。本领域常用的术语经验式是化学式,其中下标是给出一个分子中原子比率的最小整数。然而,在此,我们将“经验式”定义为成分的化学式,作为催化剂组合物中存在的极化络合剂、类盐金属和总离子氢化物的总数量比。另外,催化剂组合物被认为是催化剂聚集体的成分组合物,其中每个聚集体仅引发一个活跃的阴离子聚苯乙烯链。本发明的催化剂组合物在其成分方面是已知的,因为:(1)使用充分理解且充分表征的试剂来形成催化剂组合物;(2)相对馈料比在(a)极化络合剂的相对比率、(b)类金属盐和(c)活性金属烷基且因此存在的总离子氢化物方面明确地限定了催化剂反应混合物。然而,简单地基于已知组分的这些简单馈料比,无法知道催化剂的聚集状态。已经设计了一种简单的测试来确定催化剂聚集体组合物的平均值或在该上下文中“经验式”(如上文所定义)。该测试需要施用催化剂作为试剂作为苯乙烯(aps)在惰性无氢气氛下活跃阴离子聚合的引发剂(即,不含任何形式的链转移,实施例33-37)。然后,将所得aps分布的数均分子量(mnaps)之比与理论数均分子量mn-th相关。其中mn-th≈104*[馈入的苯乙烯摩尔数]/[形成的氢化物的总摩尔数]并且其中每摩尔所形成的氢化物等于形成氢化物的活性烷基锂和活性烷基镁的总当量(其中活性烷基锂提供一当量,并且活性烷基镁提供两当量)。为了说明该分析技术,回顾了实施例36。在实施例36中,催化剂由需要1摩尔二甲基乙醇胺、2摩尔正丁基锂和1摩尔氢气的馈料比形成。因此,该催化剂形成反应将生成具有化学式[dmea-]li2h的催化剂组合物。向该催化剂组合物中馈入苯乙烯(10.5摩尔),其相对于催化剂中存在的氢化锂的量为21摩尔,因此mn-th≈104*21≈2184道尔顿。然而,实验确定的mnaps是8447,因此mnaps/mn-th=8447/2184=3.9≈4。基于此,可以得出结论,在反应混合物中形成由[dmea-]4li8h4组成的聚集体,并且在惰性气氛下聚集体中存在氢化物中仅一个引发苯乙烯的活跃阴离子聚合。该经验式[dmea-]4li8h4与stasch通过x射线晶体学对于他的由1摩尔的本体膦酰胺配体dipnhpph2、2摩尔的仲丁基锂和苯基硅烷形成的聚集体获得的分子式[(dipnpph2)4li8h4]完全相当。因此,事实上,通过简单类比stasch的烃可溶性lih络合物和该测试方法的应用,存在有力的证据表明,经验式[dmea‾]4li8h4实际上是实施例36的催化剂组合物聚集体的至少一部分的分子式。需要指出的是,[dmea‾]4li8h4的最低整数比是[dmea‾]li2h,但是如它们用于本发明的实践,该式几乎没有带来关于催化剂聚集体组合物的信息。还要指出的是,当需要时,本发明的实践者可以使用多种极化络合剂,使得形成的聚集体包含例如[pca‾]4li8h4,其中每个[pca-]独立地相同或不同。表iii:用约1/4苯乙烯单体进料的loxlih催化剂小规模筛选实施例表v:loxlih催化剂方法,使用用在环己烷中的促进剂(即,tmeda)的全苯乙烯单体进料表vi:使用用在环己烷(mch)中的促进剂(即,tmeda)的全苯乙烯单体进料的loxlih催化剂方法表vii:loxlsh双金属催化剂方法:loxkh和loxmgh2表ix:在环己烷气氛下由以下物质制备的氢化锂引发的活跃aps分布:1)[dmea‾]8li12h4与[dmea‾]9li14h5经验催化剂组合物;或2)[dmea‾]17li22h5与[dmea‾]16li20h4经验催化剂组合物当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1