一种生物质衍生物—呋喃-2,5-二甲基氨基甲酸甲酯及其制备方法
一种生物质衍生物
—
呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯及其制备方法
技术领域
1.本发明属于有机合成领域,具体涉及一种生物质衍生物—呋喃2,5
‑
二甲基氨基甲酸甲酯及其制备方法。
背景技术:
2.氨基甲酸酯类化合物是一类具有重要生物和药物活性的含氮化合物,广泛应用于农业、医疗、卫生等领域。氨基甲酸酯也是很多天然产物的基本骨架和重要的合成中间体及化工原料,作为有机合成中间体,可用于合成异氰酸酯、杂环化合物、无毒聚氨酯、氨基甲基膦酸二苯酯、三聚氰胺衍生物、碳酸二烷基酯和聚乙烯胺等。氨基甲酸酯可以与烯烃、醛、酮、多元醇和芳环等反应,生成各种用途的衍生物,具有重要的应用价值。
3.鉴于氨基甲酸酯的应用范围甚广,因此,其合成方法受到学术界和工业界的广泛关注。传统上,氨基甲酸酯类化合物的合成主要采用以下三种方法:(1)光气法:以光气为原料,经醇解和氨解制得氨基甲酸酯;(2)氯甲酸酯法:氯甲酸酯和胺亲核取代反应得到氨基甲酸酯;(3)异氰酸酯法:直接以异氰酸酯为起始原料,与酚或醇加成反应制备氨基甲酸酯。但这些以剧毒的光气或异氰酸酯类物质为原料的合成方法因其严重腐蚀设备、污染环境、威胁工作者的安全等缺点而在工业应用中受到极大的限制。此外,市场上对氨基甲酸酯的纯度要求至少大于98.5%,尤其是医药中间体,纯度要求达到99.8%。从绿色发展战略看,以安全、可再生的原料制备高纯度氨基甲酸酯是今后化学工业发展的必然趋势。
技术实现要素:
4.本发明的目的为针对当前技术存在的不足,提供一种生物质衍生物—呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯及其制备方法。该物质具有富电子呋喃环结构,可发生卤化、硝化、磺化等亲电取代反应;此外,该物质因含有氨基甲酸酯基团,可用作农药、医药、合成树脂改性和有机合成的中间体等;所述的制备方法绿色、安全,具有良好的应用前景。
5.本发明的技术方案为:
6.一种生物质衍生物—呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯,该化合物的结构式为如下:
[0007][0008]
所述的生物质衍生物
‑
呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的制备方法,包括以下两种方法之一:
[0009]
方法一,常压反应,包括以下步骤:
[0010]
惰性气氛下,将2,5
‑
二(氨基甲基)呋喃、碳酸二甲酯和催化剂加入到常压反应器中,然后在搅拌条件下加热升温至回流温度,反应1~7小时后停止加热;待温度降至室温,向反应液中滴加盐酸酸洗至ph=1~2,然后过滤、水洗至中性,获得产物呋喃
‑
2,5
‑
二甲基
氨基甲酸甲酯;所述催化剂为甲醇钠;
[0011]
或者方法二,高压反应,包括以下步骤:
[0012]
将2,5
‑
二(氨基甲基)呋喃、碳酸二甲酯和催化剂加入到高压反应釜中,通惰性气体吹扫,然后密闭,搅拌下加热升温至60~160℃下反应1~7小时后,停止加热;待温度降至室温,放出不凝气,加热使析出的固体溶解后趁热真空过滤滤除固体催化剂,再将滤液减压蒸馏,得到产物呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯;所述催化剂为乙酸锌。
[0013]
所述2,5
‑
二(氨基甲基)呋喃和碳酸二甲酯的摩尔比为1:10~1:100;所述2,5
‑
二(氨基甲基)呋喃和催化剂的质量比为1:0.16~1:0.8。
[0014]
所述的方法一中最后水洗后得到的物质再经无水乙醇重结晶,得到纯度为99.99%以上的呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯;或方法二中,滤液减压蒸馏后得到的固体,重结晶得到纯度为99.99%以上的呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯。
[0015]
所述的惰性气体为高纯氮气或者高纯氩气。
[0016]
本发明的有益效果在于:
[0017]
1、呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯属于未见报道的新化学品,作为一种双官能团生物质衍生物,具有重要的应用价值。
[0018]
2、采用来源于生物质的2,5
‑
二(氨基甲基)呋喃和绿色化学品碳酸二甲酯为原料合成呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯,具有绿色和本质安全特征。
[0019]
3、反应条件温和,过程易于控制,目标产物收率高,且未使用贵金属催化剂,在安全性提高的同时大大降低了生产成本。收率较类似氨基甲酸酯(收率为30%左右)高,本发明的目标产物收率可达到99%,纯度可达到99.99%。
附图说明
[0020]
图1为本发明中呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的1h
‑
nmr谱图
[0021]
图2为本发明中呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的
13
c
‑
nmr谱图
[0022]
图3为本发明中呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的ms谱图
[0023]
图4为本发明中呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的ftir谱图
具体实施方式
[0024]
一、本发明的化合物的具体实施例
[0025]
实施例1
[0026]
氮气气氛下,将1.56g(即0.012摩尔)2,5
‑
二(氨基甲基)呋喃、30ml(即0.35摩尔)碳酸二甲酯和0.25g甲醇钠加入到schlenk管中。然后在搅拌条件下加热升温至85℃回流反应4小时后停止加热。待温度降至室温,向反应液中滴加17wt%盐酸溶液酸洗至ph=1~2,然后过滤,同时用蒸馏水进行水洗至中性,最后经无水乙醇重结晶获得白色晶体。经高效液相色谱分析,2,5
‑
二(氨基甲基)呋喃的转化率为100%,呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的收率为99%,纯度为99.99%。
[0027]
本实施例提供的新化合物呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的结构式如下:
[0028][0029]
本发明提供的上述化合物为白色晶体,无味,易溶于二甲基亚砜等有机溶剂,不易溶于水。分子量为242.0903,熔程为166.1℃
‑
166.7℃。
[0030]
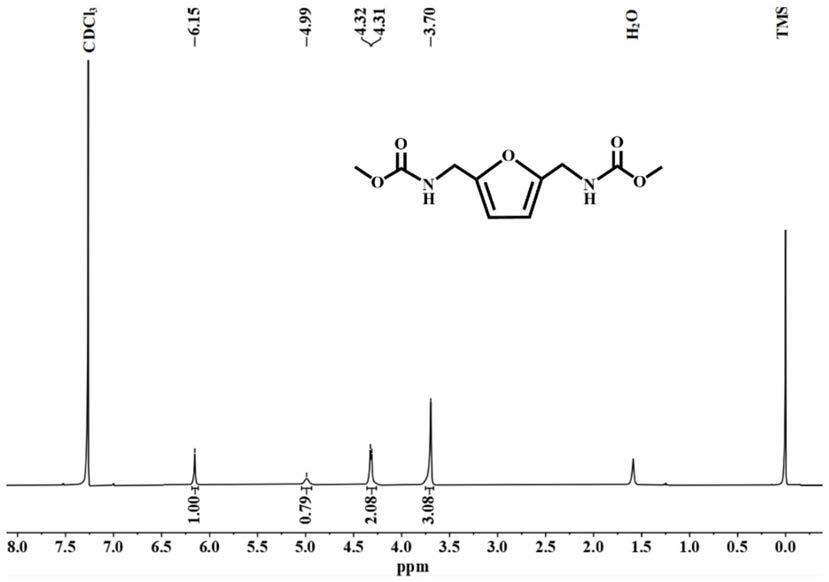
对目标化合物进行核磁分析,结果如图1、2所示,具体的:
[0031]
1)对目标化合物的氢谱数据为:1h
‑
nmr(400mhz,cdcl3):δppm 3.70(s,6h),4.31~4.32(d,j=4.0hz,4h),4.99(s,2h),6.15(s,2h);
[0032]
2)对目标化合物的碳谱数据为:
13
c
‑
nmr(400mhz,cdcl3):δppm 156.77,151.38,108.09,52.34,38.15。
[0033]
对目标化合物进行质谱分析,结果如图3所示,具体的:
[0034]
质谱图中出现m/z=243.0974[m+h]
+
,理论值243.0981;m/z=265.0793[m+na]
+
,理论值265.0800。
[0035]
对目标化合物进行红外分析,结果如图4所示,具体的:
[0036]
3255cm
‑1处吸收峰为n
‑
h键的伸缩振动;2989cm
‑1和2956cm
‑1为亚甲基伸缩振动峰;1706cm
‑1为c=o官能团的伸缩振动特征吸收峰;1683cm
‑1为仲胺弯曲振动峰;1203cm
‑1和1033cm
‑1为c
‑
o
‑
c的伸缩振动峰。
[0037]
对目标化合物进行元素分析,具体的:
[0038]
作为c
10
h
14
n2o5的理论值:c,49.58%;h,5.83%;n:11.56%。实测值:c,49.11%;h,5.77%;n:12.91%。
[0039]
结合显微熔点测定仪、1h
‑
nmr、
13
c
‑
nmr、质谱图、ft
‑
ir、以全自动元素分析仪的结果分析,证明了所合成的白色晶体为高纯度呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯。
[0040]
实施例2
[0041]
氮气气氛下,将1.56g(即0.012摩尔)2,5
‑
二(氨基甲基)呋喃、30ml(即0.35摩尔)碳酸二甲酯和1.10g甲醇钠加入到schlenk管中。然后在搅拌条件下加热升温至65℃回流反应6小时后停止加热。待温度降至室温,向反应液中滴加17wt%盐酸溶液酸洗至ph=1~2,然后过滤,同时用蒸馏水进行水洗至中性,最后经无水乙醇重结晶获得白色晶体。经高效液相色谱分析,2,5
‑
二(氨基甲基)呋喃的转化率为100%,呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的收率为98%,纯度为99.99%。
[0042]
实施例3
[0043]
将1.56g(即0.012摩尔)2,5
‑
二(氨基甲基)呋喃、30ml(即0.35摩尔)碳酸二甲酯和0.25g乙酸锌加入到高压反应釜中,通氮气吹扫,然后在搅拌条件下加热升温至160℃反应7小时后停止加热。待温度降至室温,放出不凝气,加热使析出的固体溶解后趁热真空过滤滤除固体催化剂,再将滤液减压蒸馏后得到的固体用无水乙醇重结晶获得白色晶体。经高效液相色谱分析,2,5
‑
二(氨基甲基)呋喃的转化率为100%,呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的收率为95%,纯度为99.99%。
[0044]
实施例4
[0045]
将1.0g(即0.008摩尔)2,5
‑
二(氨基甲基)呋喃、30ml(即0.35摩尔)碳酸二甲酯和0.16g乙酸锌加入到反应釜中,通氮气气体吹扫,然后在搅拌条件下加热升温至160℃反应3
小时后停止加热。待温度降至室温,放出不凝气,加热使析出的固体溶解后趁热真空过滤滤除固体催化剂,再将滤液减压蒸馏后得到的固体用无水乙醇重结晶获得白色晶体。经高效液相色谱分析,2,5
‑
二(氨基甲基)呋喃的转化率为100%,呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯的收率为92%,纯度为99.99%。
[0046]
通过以上实施例可以看出,本发明以可再生2,5
‑
二(氨基甲基)呋喃和绿色化学品碳酸二甲酯为原料制备生物质衍生物呋喃
‑
2,5
‑
二甲基氨基甲酸甲酯具有绿色、安全特征,有良好的发展前景。
[0047]
本发明未尽事宜为公知技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1