1.本发明涉及化学制药领域,具体涉及一种卡格列净杂质的制备方法。
背景技术:2.卡格列净(canagliflozin)是钠-葡萄糖共转运蛋白2(sglt2)抑制剂中获准的第一个药物,用于改善ii型糖尿病成人患者的血糖控制,该药由日本田边三菱公司和美国强生公司共同研发,2013年3月由fda批准上市。它作用于肾脏中帮助葡萄糖的重吸收的sglt2蛋白,使更多的糖通过患者的尿液排除,降低血糖水平。
3.卡格列净化学名为(1s)-1,5-脱氢-1-[3-[[5-(4-氟苯基)-2-噻吩基]甲基]-4-甲基苯基]-d-葡萄糖半水合物,其结构式如下:
[0004][0005]
文献(drug metabolism and disposition,2014,vol.42,#5,p.903
–
916)报道了卡格列净在动物以及人体内的代谢产物,其中杂质(2s,3r,4r,5s,6r)-2-(3-((5-(4-氟苯基)噻吩-2-基)甲基)-4-(羟甲基)苯基)-6-(羟甲基)四氢-2h-吡喃-3,4,5-三醇(以下简称卡格列净杂质),其结构式如化合物kgj086-32所示,为动物体内主要代谢产物。卡格列净在生产和储存过程,其结构中苯甲基易与空气中氧气发生反应,产生氧化杂质kgj086-32。因此该杂质研究对卡格列净的质量控制和杂质研究具有重要意义。
[0006][0007]
目前并无文献对该卡格列净杂质合成方法进行报道,为了提高卡格列净成品的质量,降低临床用药的风险,本发明提供了一种卡格列净杂质的制备方法,可以快速、简便、高效地得到杂质对照品,使得在质量研究中可以有效地检测和控制该杂质。
技术实现要素:[0008]
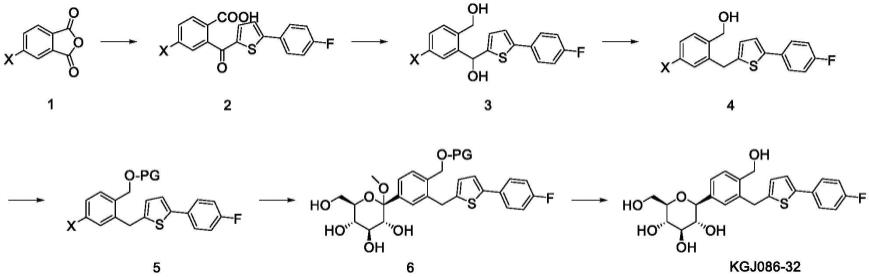
本发明提供一种化合物kgj086-32的制备方法,其包括以下步骤:
[0009][0010]
其中,x为氟、氯、溴或碘;pg为羟基保护基,例如为硅醚类保护基,如三甲基硅基、三乙基硅基、三异丙基硅基、叔丁基二甲基硅基、叔丁基二苯基硅基;
[0011]
a)化合物1与2-(4-氟苯基)噻吩在路易斯酸存在下发生傅克酰基化反应得到化合物2;
[0012]
b)所述化合物2在还原剂作用下,还原羧基和羰基反应得到化合物3;
[0013]
c)所述化合物3发生羟基还原反应得到化合物4;
[0014]
d)所述化合物4与提供保护基pg的化合物反应得到化合物5;
[0015]
e)所述化合物5与2,3,4,6-四-o-三乙基硅烷基-d-葡萄糖内酯反应得到化合物6;
[0016]
f)所述化合物6先进行脱除pg反应后,再进行脱除甲氧基反应得到化合物kgj086-32;或者,化合物6先进行脱除甲氧基反应后,再进行脱除pg反应得到化合物kgj086-32。
[0017]
根据本发明的实施方案,在步骤a)中,所述路易斯酸可以为三氯化铝、三氯化铁、三氟化硼、四氯化钛、二氯化锌中的至少一种,优选三氯化铝。
[0018]
根据本发明的实施方案,在步骤a)中,所述化合物1与路易斯酸的摩尔比为1:(1-5),例如为1:(1-3),示例性为1:1.5。
[0019]
根据本发明的实施方案,在步骤a)中,所述反应温度可以为-70~10℃,例如为:-15~10℃,示例性为0-5℃。
[0020]
根据本发明的实施方案,在步骤b)中,所述还原剂可以为硼氢化钠、硼氢化钾、硼氢化锂、四氢铝锂、硼烷二甲硫醚中的至少一种,优选为硼烷二甲硫醚。
[0021]
根据本发明的实施方案,在步骤b)中,所述化合物2与还原剂的摩尔比为1:(1-5),例如1:(1-4),示例性为1:2。
[0022]
根据本发明的实施方案,在步骤c)中,所述反应可以在硅烷和路易斯酸作用下进行,所述硅烷可以为三甲基硅烷、三乙基硅烷、三异丙基硅烷中的至少一种;所述路易斯酸选自三氯化铝、三氯化铁、三氟化硼乙醚、四氯化钛、二氯化锌中的至少一种,优选三氟化硼乙醚。
[0023]
根据本发明的实施方案,在步骤c)中,所述化合物3、硅烷和路易斯酸的摩尔比为1:(1-10):(1-10),例如为1:(2-5):(2-5),示例性为1:4:3。
[0024]
根据本发明的实施方案,在步骤c)中,所述溶剂选自二氯甲烷、乙腈、三氯甲烷、四氢呋喃、二氧六环中的至少一种,优选二氯甲烷/乙腈混合溶剂。
[0025]
根据本发明的实施方案,在步骤c)中,反应温度为-70~10℃,例如为:-50~-10℃,示例性为-30~-20℃。
[0026]
根据本发明的实施方案,在步骤d)中,所述保护基pg可以选自三甲基硅基(tms)、
三乙基硅基(tes)、三异丙基硅基(tips)、叔丁基二甲基硅基(tbs)或叔丁基二苯基硅基(tbdps);
[0027]
优选地,所述提供保护基pg的化合物可以选自三甲基氯硅烷、三乙基氯硅烷、三异丙基氯硅烷、叔丁基二甲基氯硅烷或叔丁基二苯基氯硅烷。
[0028]
根据本发明的实施方案,在步骤d)中,所述反应可以在有机碱存在下进行,所述有机碱可以为三乙胺、吡啶、4-二甲氨基吡啶、二异丙基乙胺中的至少一种;
[0029]
根据本发明的实施方案,在步骤d)中,所述化合物4、有机碱和提供保护基pg的化合物的摩尔比可以为1:(1-8):(1-5),例如1:(1-5):(1-4),示例性为1:2.5:2。
[0030]
根据本发明的实施方案,在步骤e)中,所述化合物5与2,3,4,6-四-o-三乙基硅烷基-d-葡萄糖内酯的摩尔比可以为1:(0.5-3),例如为1:(0.8-2),示例性为1:1.1。
[0031]
根据本发明的实施方案,在步骤e)中,所述反应可以在碱作用下进行,所述碱可以为有机金属试剂,例如为正丁基锂、甲基锂、二异丙基氨基锂中的至少一种。
[0032]
根据本发明的实施方案,在步骤e)中,所述化合物5与碱的摩尔比可以为1:(0.5-3),例如为1:(0.8-2),示例性为1:1.2。
[0033]
根据本发明的实施方案,在步骤e)中,所述反应可以在反应溶剂中进行,所述反应溶剂可以为四氢呋喃、甲苯和二氯甲烷中的一种、两种或更多种。
[0034]
根据本发明的实施方案,步骤e)反应结束后无需后处理可以直接进行步骤f)中所述先进行脱除pg反应后,再进行脱除甲氧基反应。
[0035]
根据本发明的实施方案,在步骤f)中,所述脱除pg反应可以在酸作用下进行,所述酸选自甲磺酸、盐酸、三氟乙酸、硫酸中的至少一种,优选甲磺酸。
[0036]
根据本发明的实施方案,在步骤f)中,所述化合物6与酸的摩尔比可以为1:(1-10),例如为1:(2-8),示例性为1:5.38。
[0037]
根据本发明的实施方案,在步骤f)中,所述脱除甲氧基反应可以在硅烷和路易斯酸作用下进行,所述硅烷可以为三甲基硅烷、三乙基硅烷、三异丙基硅烷中的至少一种;所述路易斯酸可以为三氯化铝、三氯化铁、三氟化硼、四氯化钛、二氯化锌中的至少一种。
[0038]
根据本发明的实施方案,在步骤f)中,所述化合物6、硅烷和路易斯酸的摩尔比可以为1:(1-10):(1-10),例如为1:(2-6):(2-6),示例性为1:5:4。
[0039]
根据本发明的实施方案,在步骤f)中,所述脱除甲氧基反应可以在反应溶剂中进行,所述溶剂选自二氯甲烷、乙腈、三氯甲烷、四氢呋喃,二氧六环中一种或几种,优选二氯甲烷/乙腈混合溶剂。
[0040]
根据本发明示例性的实施方案,所述kgj086-32化合物的制备方法,包括以下步骤:
[0041][0042]
a1)4-溴邻苯二甲酸酐与2-(4-氟苯基)噻吩在三氯化铝作用下发生傅克酰基化反应,得到式i化合物;
[0043]
b1)所述式i化合物经硼烷二甲硫醚还原羧酸和羰基,得到式ii化合物;
[0044]
c1)所述式ii化合物在三乙基硅烷存在下,经三氟化硼乙醚选择性还原羟基,得到式iii化合物;
[0045]
d1)所述式iii化合物与tbscl反应进行羟基保护,得到式iv化合物;
[0046]
e1)所述式iv化合物与2,3,4,6-四-o-三乙基硅烷基-d-葡萄糖内酯,在正丁基锂作用下反应,并在酸性条件下脱除保护基得到式vi化合物;
[0047]
f1)所述式vi化合物在三氟化硼乙醚和三乙基硅烷作用下,得到kgj086-32化合物。
[0048]
本发明还提供中间体化合物,具体结构如下所示:
[0049][0050]
其中,x、pg具有上文所述的定义。
[0051]
根据本发明的实施方案,化合物2-1的制备方法同化合物2;化合物3-1的制备方法同化合物3;化合物6-1和化合物6-2在所述步骤f)中得到。
[0052]
本发明还提供所述合成方法在药物工艺研究中的应用,其可用于卡格列净杂质研究。
[0053]
有益效果
[0054]
现有的技术中并未提到该杂质(未知物)及该杂质的合成方法,本发明为卡格列净
杂质(kgj086-32)的合成提供了新思路,本发明提供的中间体化合物和kgj086-32化合物的合成方法起始物料易得、操作简单,收率较高,能够满足对该杂质的制备需求,对卡格列净杂质的研究具有重要意义。
附图说明
[0055]
图1为实施例2中式ii化合物纯品的1h-nmr谱图,溶剂为氘代dmso;
[0056]
图2为实施例2中式ii化合物纯品的noe谱图;
[0057]
图3为实施例5中tlc监测图;
[0058]
图4为实施例6中kgj086-32纯品的1h-nmr谱图,溶剂为氘代dmso;
[0059]
图5为实施例6中kgj086-32纯品的液质谱图;
[0060]
图6为实施例6中kgj086-32纯品的hplc液相谱图。
具体实施方式
[0061]
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0062]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0063]
实施例1式i化合物的合成
[0064][0065]
将4-溴邻苯二甲酸酐(20g,1eq),dcm(600ml)加入到1l三口瓶中搅拌溶清,加入无水三氯化铝(17.6g,1.5eq),氩气置换三次,降温至0-5℃左右,分批加入2-(4-氟苯基)噻吩(17.2g,1.1eq),氩气置换三次,保温反应12h。原料反应完,缓慢滴加入600ml水淬灭,分液,有机相用饱和氯化钠洗一次,无水硫酸钠干燥,抽滤,减压浓缩至少量,搅拌均匀,过滤,烘干,得式i和式i-1混合物,浅黄色固体32g,收率:89.9%。
[0066]
实施例2式ii化合物的合成
[0067][0068]
取实施例1中得到的混合物(32g,1eq),thf(150ml)加入到500ml三口瓶中搅拌溶清,降温至0-5℃左右,滴加硼烷二甲硫醚溶液(2eq)。滴完后保温0℃左右反应4小时。滴加甲醇淬灭反应,减压浓缩干,残留物用硅胶柱层析(200-300目)纯化,流动相石油醚/乙酸乙酯=3:1分离,减压浓缩干式ii化合物13.3g,收率:42.9%,检测谱图见图1-2。
[0069]
实施例3式iii化合物的合成
[0070][0071]
取实施例2中得到的式ii化合物(13.3g,1eq)用二氯甲烷(270ml)、乙腈(270ml)溶解于1l三口瓶中,氩气置换三次,液氮-乙醇降温至-30~-20℃以下,依次滴加三乙基硅烷(15.7g,4eq)、三氟化硼乙醚(14.5g,3eq)。滴完后保温反应过夜。原料反应完,加入600ml水,分液,有机相饱和氯化钠洗一次,无水硫酸钠干燥,抽滤,减压浓缩干,残留物用硅胶柱层析(200-300目)纯化,流动相石油醚/乙酸乙酯=3:1分离,减压浓缩干得纯品式iii化合物11.5g,收率90%。
[0072]
实施例4式iv化合物的合成
[0073][0074]
取实施例3中得到的式iii化合物(11.5g,1eq),dcm(115ml),三乙胺(7.7g,2.5eq)加入到500ml三口瓶中,降温至0℃,分批加入tbscl(9.2g,2eq),升室温反应过夜12h。原料反应完,加入300ml水,200ml二氯甲烷,分液,有机相饱和氯化钠洗一次,无水硫酸钠干燥,抽滤,减压浓缩干,残留物用硅胶柱层析(200-300目)纯化,纯石油醚流动相分离,减压浓缩干得式iv化合物14.5g,收率:96.6%。
[0075]
实施例5式vi化合物的合成
[0076][0077]
取实施例4中得到的式iv化合物(15.0g,1eq),thf(120ml)加入到500ml三口瓶中,氩气置换三次,液氮-乙醇降温至-80℃以下,滴加正丁基锂(14.7ml)。滴完后保温在-80℃左右搅拌半小时。滴加2,3,4,6-四-o-三乙基硅烷基-d-葡萄糖内酯(15.7g,1.1eq)的thf(30ml)溶液,滴完后保温在-80℃左右搅拌反应3小时。tlc监测如图3(展开剂,pe:ea=15:1)观察到中间态,保温滴加甲磺酸(15g)的甲醇(100ml)溶液,滴完后升室温反应过夜。中间态反应完,加入500ml水,用乙酸乙酯萃取2次,有机相饱和氯化钠洗一次,无水硫酸钠干燥,抽滤,减压浓缩干,残留物用硅胶柱层析(200-300目)纯化,流动相二氯甲烷/甲醇=10:1分离,减压浓缩干共得式vi化合物7.9g,收率:52.7%。
[0078]
实施例6化合物kgj086-32的合成
[0079][0080]
取实施例5中得到的式vi化合物(5.8g,1eq)用二氯甲烷(116ml)、乙腈(116ml)溶解于500ml三口瓶中,氩气置换三次,液氮-乙醇降温至-30℃以下,依次滴加三乙基硅烷(6.9g,5eq)、三氟化硼乙醚(6.7g,4eq)。滴完后升室温反应过夜。lcms监测原料反应完,加入300ml水,用二氯甲烷萃取4次,有机相无水硫酸钠干燥,抽滤,减压浓缩干,残留物用硅胶柱层析(200-300目)纯化,流动相二氯甲烷/甲醇=10:1分离,减压浓缩干得4.7g。用水打浆,过滤,固体冻干机冻干,得kgj086-32化合物4.0g。hplc:98.75%,收率:86.4%,检测谱图见图4-6。
[0081]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。