一种苯溴马隆关键中间体的合成方法与流程
本发明涉及生物医药技术领域,具体涉及一种苯溴马隆关键中间体的合成方法。
背景技术:
苯溴马隆(benzbromarone),商品名痛风利仙,化学名为((3,5-二溴-4-羟基苯基)-(2-乙基-3-苯骈呋喃基)甲酮),其结构如式(i)所示,是一种新型的尿酸排泄促进剂,它可以改善血液中尿酸过高所引起的疾病如痛风及伴有高尿酸血症的高血压等。文献中报道的苯溴马隆的合成方法如下:(1)第一步傅克酰基化反应,使用四氯化锡或三氯化铝作为催化剂,以二氯甲烷或氯仿作为反应溶剂;(2)第二步使用盐酸吡啶进行去甲基化反应;(3)第三步采用溴素进行溴代反应。
现有的苯溴马隆合成方法仍然存在部分步骤反应能耗较高的问题,在环保要求日益提高的当前显得尤为突出,收率也仍然有改进的空间。
技术实现要素:
本发明要解决的技术问题是:提供一种反应条件温和、收率更高的苯溴马隆关键中间体的合成方法。
本发明解决上述技术问题的技术方案如下:
一种苯溴马隆关键中间体的合成方法,包括如下步骤:
(1)2-乙基苯并呋喃、对甲氧基苯甲酰氯在三氯化铝的催化下,以甲苯为溶剂发生傅克酰基化反应生成2-乙基-3-对甲氧基苯甲酰-苯并呋喃;
(2)2-乙基-3-对甲氧基苯甲酰-苯并呋喃与三氯化铝,以甲苯为溶剂进行去甲基化反应得到苯溴马隆关键中间体2-乙基-3-对羟基苯甲酰-苯并呋喃;具体反应式如下:
优选的,步骤(1)中所述2-乙基苯并呋喃、对甲氧基苯甲酰氯与三氯化铝的摩尔比为1:1~2:1~3。
进一步的,步骤(1)中所述2-乙基苯并呋喃、对甲氧基苯甲酰氯与三氯化铝的摩尔比为1:1.01~1.1:1.01~1.2。
优选的,所述步骤(1)中所述甲苯的添加质量为2-乙基苯并呋喃的2~10倍。
进一步的,所述步骤(1)中所述甲苯的添加质量为2-乙基苯并呋喃的3~6倍。
优选的,所述步骤(1)还包括如下后处理步骤:加入稀盐酸除去多余的三氯化铝,反应液分层后,甲苯提取水相,合并有机相后浓缩至干,加入乙醇水溶液溶解杂质,过滤得到的固体即为4-甲氧基苯基-(2-乙基-3-苯骈呋喃基)甲酮,可直接投入下一步反应。
优选的,所述步骤(2)中2-乙基-3-对甲氧基苯甲酰-苯并呋喃与三氯化铝的摩尔比为1:3~5。
进一步的,所述步骤(2)中2-乙基-3-对甲氧基苯甲酰-苯并呋喃与三氯化铝的摩尔比为1:3.5~4。
优选的,所述步骤(2)中甲苯的添加质量为2-乙基-3-对甲氧基苯甲酰-苯并呋喃的4~10倍。
进一步的,所述步骤(2)中甲苯的添加质量为2-乙基-3-对甲氧基苯甲酰-苯并呋喃的4~8倍。
优选的,所述步骤(2)中反应温度为0~72℃。
优选的,所述步骤(2)还包括如下后处理步骤:入稀盐酸除去多余的三氯化铝,反应液分层后,甲苯提取水相,合并有机相后浓缩、降温结晶得到4-羟基苯基-(2-乙基-3-苯并呋喃基)甲酮。
进一步的,所述步骤(2)后处理步骤中合并有机相后浓缩至4-甲氧基苯基-(2-乙基-3-苯骈呋喃基)甲酮投料质量的1.0~1.2倍。发明人发现,以甲苯为反应溶剂并在后处理中作为重结晶溶剂,能够明显提高反应的收率和直接得到产品的纯度。
本发明中化合物的中文命名与结构式有冲突的,以结构式为准;结构式有明显错误的除外。
本发明提供的苯溴马隆关键中间体的合成方法反应条件温和,选用甲苯作为两步反应的溶剂,得到的产品收率和直接纯度均较高,具有良好的工业化生产前景。
附图说明
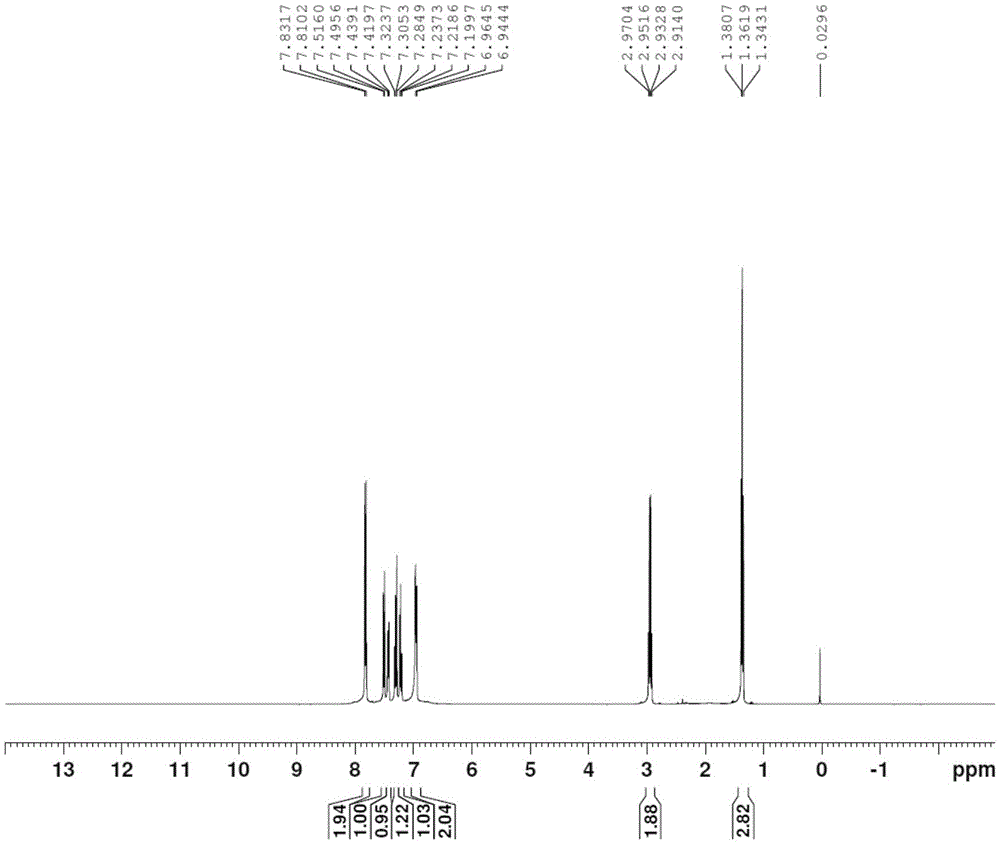
图1为本发明具体实施方式得到的苯溴马隆关键中间体2-乙基-3-对羟基苯甲酰-苯并呋喃氢谱图。
具体实施方式
以下结合实例说明本发明,但不限制本发明。在本领域内,技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案内。
实施例1:
将甲苯250g、2-乙基苯并呋喃(50g,0.342mol)、对甲氧基苯甲酰氯(65g,0.381mol)依次投入1l反应瓶中,机械搅拌下将反应体系冷却至0~5℃,开始往反应瓶中分批加入氯化铝(50g,0.375mol),投料过程中温度不超过10℃,投料完毕后保持内温在10~15℃,继续反应1~2小时,tlc监控反应进程(展开剂为pe:ea=3:1),待2-乙基苯并呋喃基本消失,反应完毕。
将10%的稀盐酸500ml投入1l反应瓶中,缓慢往反应瓶中滴加上述反应液,滴加完毕,将反应体系保持在60-65℃,搅拌1-2小时,静置分层;用50g甲苯萃取水相,后合并有机相,用500ml水洗涤有机相至中性。浓缩有机相至干,加入75%乙醇100g,将反应体系升温至55-60℃搅拌1~2h,降温至0~5℃后过滤,滤饼干燥得到黄色固体95.11g(理论收率95.81g),摩尔收率99.27%,无需纯化可直接投入下一步反应。
将甲苯350g、上一步反应的产物4-甲氧基苯基-(2-乙基-3-苯骈呋喃基)甲酮(72g,0.257mol)依次投入1l反应瓶中,将反应体系冷却至5-10℃,搅拌30分钟,开始往反应瓶中分批加入氯化铝(120g,0.9mol),投料过程中温度不超过10℃,投料完毕后保持内温在10-15℃,保温搅拌20min后升温至50-60℃,继续反应至tlc监控反应进程,待原料基本消失,反应完毕。
将10%的稀盐酸720ml投入2l反应瓶中,缓慢往反应瓶中滴加上述反应液,滴加过程中温度不超过70℃,滴加完毕搅拌10-30min,静置分层,甲苯萃取水相后合并有机相,有机相水洗两次至中性。浓缩有机相至4-甲氧基苯基-(2-乙基-3-苯骈呋喃基)甲酮投料质量的1.2倍,降温到0-5℃结晶,过滤后得到的滤饼干燥得到土黄色固体67.34g(理论产量68.40g),经hplc检测纯度为98.72%,摩尔收率97.20%。
实施例2:
将甲苯250g、2-乙基苯并呋喃(50g,0.342mol)、对甲氧基苯甲酰氯(59g,0.345mol)依次投入1l反应瓶中,机械搅拌下将反应体系冷却至0~5℃,开始往反应瓶中分批加入三氯化铝(46.7g,0.350mol),投料过程中温度不超过10℃,投料完毕后保持内温在10~15℃,继续反应1~2小时,tlc监控反应进程(展开剂为pe:ea=5:1),待2-乙基苯并呋喃基本消失,反应完毕。
将10%的稀盐酸500ml投入1l反应瓶中,缓慢往反应瓶中滴加上述反应液,滴加完毕,将反应体系保持在60-65℃,搅拌1-2小时,静置分层;用50g甲苯萃取水相,后合并有机相,用500ml水洗涤2次至中性。浓缩有机相至干,加入75%乙醇100g,将反应体系升温至55-60℃搅拌1~2h,降温至0~5℃后过滤,滤饼干燥得到黄色固体95.34g(理论收率95.81g),摩尔收率99.51%,无需纯化可直接投入下一步反应。
将甲苯350g、上一步反应的产物4-甲氧基苯基-(2-乙基-3-苯骈呋喃基)甲酮(72g,0.257mol)依次投入1l反应瓶中,将反应体系冷却至5-10℃,搅拌30分钟,开始往反应瓶中分批加入氯化铝(120g,0.9mol),投料过程中温度不超过10℃,投料完毕后保持内温在10-15℃,保温搅拌20min后升温至50-60℃,继续反应至tlc监控反应进程,待原料基本消失,反应完毕。
将10%的稀盐酸720ml投入2l反应瓶中,缓慢往反应瓶中滴加上述反应液,滴加过程中温度不超过70℃,滴加完毕搅拌10-30min,静置分层,甲苯萃取水相后合并有机相,有机相水洗两次至中性。浓缩有机相至4-甲氧基苯基-(2-乙基-3-苯骈呋喃基)甲酮投料质量的1.0倍,降温到0-5℃结晶,过滤后得到的滤饼干燥得到土黄色固体67.75g(理论产量68.40g),经hplc检测纯度为98.32%,摩尔收率97.39%。
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!