一种羟基氧杂环烷烃衍生物的制备方法与流程
本发明属于医药化工
技术领域:
,涉及一种羟基氧杂环烷烃衍生物的连续制备方法,特别涉及烯醇衍生物通过环氧化、分子内醇解关环连续制备羟基氧杂环烷烃衍生物的方法。
背景技术:
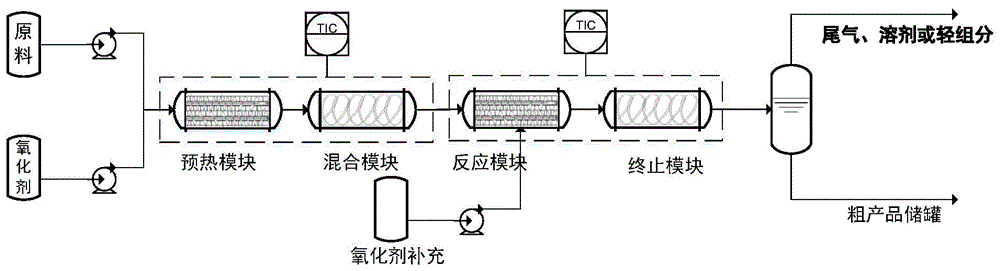
:3-羟基四氢呋喃是一种重要的医药化工中间体,在抗艾滋病药、抗癌药、降糖药等药物的生产中应用广泛。目前,3-羟基四氢呋喃主要用化学法合成,如用苹果酸(苹果酸酯、苹果酸还原产物1,2,4-丁三醇)和酒石酸(酒石酸酯)为起始原料通过酯化、还原、脱水环合合成3-羟基四氢呋喃及其手性体(s)-3-羟基四氢呋喃及(r)-3-羟基四氢呋喃(j.am.chem.soc.,1958,80,364;cn101367780a,cn104478833a,j.org.chem.,1983,48,2767;us2011/118511)。李勇智等报道了以(s)-4-氯-3-羟基丁酸乙酯为原料,经过还原和环化两步反应得到目标产物(s)-3-羟基四氢呋喃,反应总收率为75.2%(应用化工,2008,037,191)。bats等报道了一种利用2-环氧乙烷基乙醇环合制备3-羟基四氢呋喃的方法,产物中同时存在大量的2-羟甲基氧杂环丁烷(tetrahedron,1982,38,2139),但该方法所用的原料难以制备。王剑峰等报道了用小分子催化不对称合成(s)-3-羟基四氢呋喃,以4-氯丁醛和亚硝基苯为原料,经氨氧化反应、硼氢化钠还原、分子内环合制备(安普那韦中间体的合成工艺研究[d].浙江工业大学,2011)。另外一些方法利用二氢呋喃为原料制备3-羟基四氢呋喃。以二氢呋喃为原料,通过硅氢化或者硼氢化还原方法也可制得3-羟基四氢呋喃。brown等用手性硼催化剂实现了2,3-二氢呋喃和2,5-二氢呋喃的不对称硼氢化还原,产物3-羟基四氢呋喃的收率达到92%(j.am.chem.soc.,1986,108,2049)。hayashi等报道了以2,5-二氢呋喃为原料通过不对称硅氢化合成3-羟基四氢呋喃的方法(tetrahedronlett,.1993,34,2335)。目前的报道的路线方法大都存在着原料昂贵,过程复杂,或者收率不高等问题。技术实现要素:针对上述现有技术存在的问题,本公开的发明人经过多次实验发现,使用特定的原料和工艺,在特定的反应参数和反应步骤下,可以简化反应流程,缩短反应时间,并在此基础上完成了本公开。本公开的一个目的在于提供一种具有结构的羟基氧杂环烷烃衍生物(化合物(b))的制备方法,其特征在于,包括以下步骤:1)将起始反应原料化合物(a)与催化剂一起用溶剂配成原料质量百分比浓度为5-80%的原料液,与氧化物料分别从不同物料输送设备一起泵入连续流反应器预热模块进行预热,预热温度为-10-200℃;2)经过预热模块的物料进入混合模块,混合后的物料进入反应模块,反应模块温度为-10-220℃,在反应模块连续反应0.05-24小时得到反应混合物;3)在反应模块反应后,反应混合物进入产品分离模块,将反应液在冷却、或反应温度下、或在提升的温度下经过有机无机分离或经过溶剂脱除得到粗产品(b)粗产品经过精制得到纯品。其中,r1、r2、r3、r4各自独立地是氢、c1-c10的烷基、氨基、卤素取代的c1-c10烷基,n为1至3的整数。有益效果根据本公开的使用特定的原料和工艺,在特定的反应参数和反应步骤下,可以简化反应流程,缩短反应时间,更高效地合成羟基氧杂环烷烃衍生物。附图说明图1为根据本申请的一个实施方式使用的微结构反应器的示意图。具体实施方式为使本领域具有普通知识的人员可了解本发明的特点及效果,以下谨就说明书及申请专利范围中提及的术语及用语进行一般性的说明及定义。除非另有指明,否则文中使用的所有技术及科学上的字词,皆具有本领域技术人员对于本发明所了解的通常意义,当有冲突情形时,应以本说明书的定义为准。以下具体实施方式本质上仅是例示性,且并不欲限制本发明及其用途。此外,本文并不受前述现有技术或
发明内容或以下具体实施方式或实施例中所描述的任何理论的限制。根据本公开的一个实施方式,其提供了一种具有结构的化合物(b)的制备方法,其特征在于,包括以下步骤:1)将起始反应原料(a)与催化剂用溶剂配成原料浓度为5-80%的原料液,与氧化物料分别从不同物料输送设备一起泵入连续流反应器预热模块进行预热,预热温度为-10-200℃;2)经过预热模块的物料进入混合模块,混合后的物料进入反应模块,反应模块温度为-10-220℃,在反应模块连续反应0.05-24小时得到反应混合物;3)在反应模块反应后,反应混合物进入产品分离模块,将反应液在冷却、或反应温度下、或在提升的温度下经过有机无机分离或经过溶剂脱除得到粗产品(b)粗产品经过精制得到纯品。其中,在化合物(a)和(b)中r1、r2、r3、r4各自独立地是氢,c1-c10的烷基,氨基,卤素取代的c1-c10烷基,n为1、2或3的整数。优选地,r1、r2、r3、r4各自独立地是氢,c1-c4的烷基,氨基,卤素取代的c1-c4烷基。优选地,所述卤素选自f、cl和br。进一步优选地,r1、r2、r3、r4各自独立地是氢。优选地,n为1或2的整数,进一步优选地,n为1。根据本公开的一个实施方式,其中,步骤1)中所述的溶剂为二氯甲烷、二氯乙烷、氯仿、苯、氯苯、乙醚、四氢呋喃、二氧六环、乙腈、甲苯、水、甲醇、乙醇、异丙醇、甲酸、乙酸、烷烃、环烷烃的一种或者其混合物;优选为二氯甲烷、二氯乙烷、四氢呋喃、乙腈、氯仿、或氯苯,更优选为二氯甲烷或二氯乙烷。通过使用上述特定溶剂,可以更好地控制传热传质过程和流体的流速,从而使反应顺利进行。而且,在一些实施方案中,催化剂的溶剂分解作用明显,采用质子型溶剂将变得不合适。质子溶剂在催化剂的作用下与烯醇环氧化形成的中间产物2-(环氧乙烷-2-基)乙-1-醇醇解,水解或者溶解分解作用(如醇解反应),因此可以根据催化剂分散形态优化溶剂选择,优选地,选择二氯甲烷、二氯乙烷、四氢呋喃、乙腈、氯仿、氯苯作为反应溶剂。一方面可以促进反应快速进行(环氧化反应在氯仿、氯苯或二氯甲烷中反应时有较高反应速率),另一方面,可以防止中间产物的溶剂分解作用。根据本公开的一个实施方式,其中,所述步骤1)使用的催化剂为选自无机酸、有机酸、或杂多酸、磷钨酸、乙酰丙酮氧钒、乙酰丙酮氧钨、乙酰丙酮氧钛、乙酰丙酮氧钼或者其组合。优选为磷钨酸、乙酰丙酮氧钒、乙酰丙酮氧钨、乙酰丙酮氧钛和乙酰丙酮氧钼中的一种或多种。更优选为磷钨酸和/或乙酰丙酮氧钒。所述催化剂的用量为原料化合物(a)重量的0.15%~20%。当催化剂的用量低于0.15%时,反应效果太低;当催化剂的用量大于20%时,副反应产物增加。优选地,所述催化剂的用量为原料化合物(a)重量的0.2-3%,更优选为0.2-1.0%,在此范围时,反应效率最优。通过使用上述特定的催化剂及特定的用量,可以有效地控制反应速度,使反应顺利进行,并提高反应的安全系数。根据本公开的一个实施方式,其中,所述步骤1)使用的氧化物料为氧化剂与有机酸的混合水溶液或混合物。所述氧化剂选自过氧化叔丁醇、过氧苯甲酸、间氯过氧苯甲酸、三氟过氧乙酸、过氧甲酸、过氧乙酸、二甲基过氧化酮、甲基三氟甲基二环氧乙烷、过硫酸氢钾、过氧化氢、异丙苯过氧化氢、和氧气中的一种或多种。虽然上述多种氧化剂均可实现本公开的氧化效果,但考虑到安全性和经济性,优选的氧化剂为过氧化氢。所述有机酸选自甲酸、乙酸或丙酸,更优选为甲酸。有机酸的使用可以提高氧化反应的效率,例如在其它反应条件相同的情况下,氧化剂用量更少;或者双氧水和甲酸或者乙酸一起混合处理,可以得到部分过氧甲酸、过氧乙酸,氧化效果更好。氧化剂与原料化合物(a)的摩尔比例为1:0.1-1:100,优选为1:1-1:50,更优选为1:1.1-1:10。通过使用上述特定的氧化剂,可以有效地控制反应速度,使反应顺利进行,并提高反应的安全系数。根据本公开的一个实施方式,优选地,所述步骤1)中的预热温度为10-150℃。根据本公开的一个实施方式,优选地,所述步骤2)中的反应模块温度为50-120℃。在所述特定的反应温度下,可以有效地平衡反应热,提高反应的安全系数。根据本公开的一个实施方式,优选地,所述步骤2)中的反应时间为10min至50min。根据本公开的一个实施方式,其中,该方法使用微结构连续反应器进行。微结构连续反应器(微通道、连续流)在反应器的设计上增强和优化反应器的传质和传热性能,可以起到过程强化的作用。而且,环氧化反应涉及过氧酸环氧化过程,存在一定的安全风险,用微通道连续流方式进行环氧化过程,可以通过高效的传质、传热能力有效控制体系温度变化,利用灵活的反应参数调整(例如,流体流速、反应模块持液时间),监控反应进度。根据本公开的一个实施方式,其中,所述反应器的预热模块、混合模块、反应模块和产品分离模块可以为四个独立模块或者任意两个、三个或者四个连续单元为同一组件模块。通过这样的模块化设计,可以简化反应设备。通过这些模块组合可以完成多步反应,且不同反应单元模块易于调整,起到节能减排的作用。根据本公开的一个实施方式,其中,所述反应模块中,持液单元通道为回型、八卦形、心型、伞型、s型,z型、o型、螺旋型、中空型或它们的组合;反应模块中,持液单元通道通过静态组合或者配有混合动力单元。通过这些具有特定形状的持液单元通道的反应模块和配有混合动力单元,可以进一步地增强和优化反应器的传质和传热性能,起到过程强化的作用,从而促进反应顺利进行。根据本公开的一个实施方式,其中,在所述反应模块中,持液单元的物料体积为20ml-50000ml。在上述特定工艺条件下,可以控制反应进程,提高产物收率,促进反应顺利进行。根据本公开的一个实施方式,其中,所述化合物(a)为3-丁烯-1-醇,化合物(b)为3-羟基四氢呋喃。根据本发明采用从3-丁烯-1-醇环氧耦合分子内关环路线连续制备3-羟基四氢呋喃的路线,相比于现行的1,2,4-丁三醇原料更加经济,过程更高价安全,操作简单,有利于大规模工业化生产。相比于asimbhaumik等报道的一种钛硅分子筛催化丁烯醇环氧化耦合环合制备3-羟基四氢呋喃的方法(chem.commun.,1998463),本发明所述的方法采用连续的反应技术,大大降低了间歇环氧化过程的安全风险,同时,借助连续反应方式,传质过程大大强化,反应时间进一步缩短。此外,根据本公开的一个实施方式,也可以使用固体催化剂,如ti-mww,ts-1,ti-betaandti-mor。但在此情况下,如果反应器的孔道尺寸较小(如<3mm),则基于反应液体质量,所述固体催化剂的使用量不高于5%。如果反应器的孔道尺寸较大,催化剂用量可以提高以促进反应进度和提高反应效率;尤其在一些特定具有输送能力单元的连续反应器中,如螺旋输送、双螺旋挤压输送反应器中,固体催化剂的用量可以提升到20%以上,从而可以使烯醇原料高效转化。3-羟基四氢呋喃可以作为前驱体用于进一步转化为可以合成药物直接使用的手性3-羟基四氢呋喃,这种转化方法是已知的,例如cn201510572955.9,j.am.chem.soc.2012,acscatal.,2016,1598;febsjournal2013,280,3084-3093中公开的技术。实施例以下实施例仅是作为本发明的实施方案的例子列举,并不对本发明构成任何限制,本领域技术人员可以理解在不偏离本发明的实质和构思的范围内的修改均落入本发明的保护范围。实施例1微通道反应器选用山东豪迈化工技术有限公司cs200型碳化硅微通道,持液体积为100ml,配备双柱塞泵进料,将高低温一体循环机温度设定为50℃。将3-丁烯-1-醇用二氯甲烷配成浓度为20%的原料溶液,然后加入磷钨酸催化剂,磷钨酸的加入量为3-丁烯-1-醇的0.2%;将质量分数为30%双氧水、甲酸、蒸馏水按体积比为5:4:1的比例配成氧化物料;将原料流和氧化物料按摩尔比为1:1.5分别通过普罗名特浆料泵和高压恒流柱塞计量泵注入cs200型微通道反应器中,总液体流速为2ml/min,在微反应器中可停留50min,在出口处用盘管冷却,经过亚硫酸钠洗涤后进行气相分析。通过产物在色谱上的保留时间对产物进行定性分析,该分析在配备了自动进样器aoc-20的岛津2010plus气相色谱上进行。气相色谱定量条件:色谱柱选用cp-wax58(ffap,25m×0.25mm×0.2μm,chrompack);汽化室温度250℃(分流比1:30);fid检测器温度280℃;柱温箱温度在60℃保持1min,然后以20℃/min的速率升至250℃并保持5min;气路控制:n21ml/min(column),h230ml/min,空气300ml/min,尾吹n229ml/min。结果显示,当没有催化剂使用时(空白对照),主要产物为环氧化产物2-(环氧乙烷-2-基)乙-1-醇,仅有少量3-羟基四氢呋喃产品;在本实施例1的考察的条件下,3-羟基四氢呋喃收率可达到32%。实施例2-6按照实施例1中相同的方式进行试验,不同之处在于通过改变下表1中列出的不同反应条件,主要为反应停留时间和反应温度,结果如下表1所示。表1实施例温度催化剂用量总流速停留时间转化率收率空白对照5002.0ml/min50min76%2%1500.2%2.0ml/min50min76%32%2500.5%3.5ml/min28.6min80%53%3800.5%5.0ml/min20min96%65%4800.5%6.0ml/min16.6min98%76%51001.0%8.5ml/min11.76min99%81%61200.5%6.8ml/min14.7min100%83%从表1的数据可以看出,在优化的反应条件120℃,催化剂用量0.5%,总流速6.8ml/min,反应停留时间14.7min,原料转化率可达100%,目标产物3-羟基四氢呋喃的收率可达83%。实施例7微通道反应器选用山东豪迈化工技术有限公司cs200型碳化硅微通道,持液体积为100ml,配备双柱塞泵进料,将高低温一体循环机温度设定为80℃。将3-丁烯-1-醇用二氯甲烷配成浓度为20%的原料溶液,然后加入乙酰丙酮氧钒催化剂,乙酰丙酮氧钒的加入量为3-丁烯-1-醇的0.5%;将质量分数为30%双氧水、甲酸、蒸馏水按体积比为4:4:2的比例配成氧化物料;将原料流和氧化物料按一定的体积流速比(控制氧化剂与原料摩尔比为1:3分别通过普罗名特浆料泵和高压恒流柱塞计量泵注入cs200型微通道反应器中,在微反应器中以5.0ml/min进料,停留20min后,在出口处用盘管冷却接,经过亚硫酸钠洗涤后进行气相分析。结果显示,3-羟基四氢呋喃收率可达到46%。实施例8-10按照实施例7中相同的方式进行试验,不同之处在于通过改变下表2中列出的不同反应条件,主要为反应停留时间和反应温度,结果如下表2所示。表2实施例温度催化剂用量总流速停留时间转化率收率7800.5%5.0ml/min20min100%46%8801.0%5.0ml/min20min100%68%91001.0%8.5ml/min11.76min100%72%101200.5%6.8ml/min14.7min100%66%实施例11微通道反应器选用自制3mm(内径为1.5mm)盘管,总持液体积为600ml,配备双柱塞泵进料,将3mm盘管放入油浴锅中,温度设定为80℃。将3-丁烯-1-醇用二氯甲烷配成浓度为20%的原料溶液,然后加入乙酰丙酮氧钒催化剂,乙酰丙酮氧钒的加入量为3-丁烯-1-醇的0.5%;将质量分数为30%双氧水、甲酸、蒸馏水按体积比为5:4:1的比例配成氧化物料;将原料流和氧化物料按一定的体积流速比(控制氧化剂与原料摩尔比为1:5分别通过普罗名特浆料泵和高压恒流柱塞计量泵注入盘管中,以30ml/min进料,在微反应器中停留20min后,在出口处用盘管冷却接,经过亚硫酸钠洗涤后进行气相分析,结果显示,产品3-羟基四氢呋喃收率可达43%。实施例12-14按照实施例11中相同的方式进行试验,不同之处在于通过改变下表2中列出的不同反应条件,主要为反应停留时间和反应温度,结果如下表3所示。表3实施例温度催化剂用量总流速停留时间转化率收率11800.5%30ml/min20min100%43%12801.0%30ml/min20min100%51%131001.0%40ml/min15min100%72%141200.5%40ml/min15min100%80%从表3的数据可以看出,在优化的反应条件120℃,催化剂用量0.5%,总流速40ml/min(2.4l/h),反应停留时间15min,原料转化率可达100%,目标产物3-羟基四氢呋喃的收率可达80%。以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本
技术领域:
的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应所述以权利要求的保护范围为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1