一种多环稠合的1,5-苯并二氮杂卓化合物及其制备方法
一种多环稠合的1,5
‑
苯并二氮杂卓化合物及其制备方法
技术领域
1.本发明涉及化学合成技术领域,尤其涉及一种多环稠合的1,5
‑
苯并二氮杂卓化合物及其制备方法。
背景技术:
2.1,5
‑
苯并二氮杂卓是一类具有“特权结构”的重要苯并七元含氮杂环化合物,是许多天然产物和药物分子的骨架,具有多种生物活性,是新药研发中一类具有价值的先导化合物。最具代表性的药物如氯氮平、奥氮平等,已作为治疗精神分裂症和癫痫的特效药应用于临床。研究人员还发现,1,5
‑
苯并二氮杂卓类化合物在癌症、心血管疾病等具有较好的疗效,有些还具有抑制细菌和真菌的活性。更令人关注的是,多环拼合或稠合的1,5
‑
苯并二氮杂卓类化合物对艾滋病的hiv
‑
1病毒蛋白酶和丙型肝炎病毒(hcv)具有较强的抑制作用,因此,多环稠合的1,5
‑
苯并二氮杂卓是一类重要的药物分子。
3.因其在医学领域和工业生产上都具有广泛的应用前景,1,5
‑
苯并二氮杂卓化合物的设计与合成受到越来越多科研工作者的关注,因此,合成具有新型结构的1,5
‑
苯并二氮杂卓化合物对于发展抗焦虑、抗惊厥、抗真菌药物、抗癌或抗艾滋病具有十分重要的意义。且目前现有的合成1,5
‑
苯并二氮杂卓类化合物的方法较为繁琐,无法实现工业化生产。因此,也需要研发一种新颖、温和的绿色方法来合成重要的1,5
‑
苯并二氮杂卓类化合物。
技术实现要素:
4.为了丰富1,5
‑
苯并二氮杂卓类化合物的种类以及解决现有技术中合成1,5
‑
苯并二氮杂卓化合物的方法较为复杂的问题,本发明提供一种多环稠合的1,5
‑
苯并二氮杂卓化合物及其制备方法。
5.为解决上述技术问题,本发明提供的技术方案是:
6.一方面,本发明提供了一种多环稠合的1,5
‑
苯并二氮杂卓化合物,其结构如式(i)或式(ⅱ)所示:
[0007][0008]
其中,r1、r2为h、甲基、乙基、丙基或卤素;
[0009]
r3为h、甲基或乙基;r4为甲基、乙基或丙基;
[0010]
n为1或2。
[0011]
相对于现有技术,本发明提供了一种新型结构的1,5
‑
苯并二氮杂卓化合物,其是
一种多环稠合的苯并二氮杂卓化合物,并在其结构上引入了烷氧羰基甲基(
‑
ch2coor),
‑
ch2coor是一种药物活性基团,引入到苯并二氮杂卓化合物的结构后会增加其在生物体内的脂溶性,从而增加其药理活性,因此,本发明不但丰富了1,5
‑
苯并二氮杂卓化合物的种类,还为发展抗艾滋病或丙型肝炎药物提供了一种新化合物,对于研究该类化合物的活性以及扩大该类化合物在医学领域和工业生产上的应用具有十分重要的意义,为研究具有高生理活性的新型药物提供了基础。
[0012]
可选的,所述卤素为cl或br。
[0013]
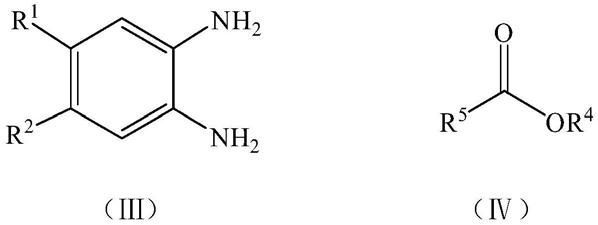
另一方面,本发明还提供了一种上述多环稠合的1,5
‑
苯并二氮杂卓化合物的制备方法,以式(iii)所示的取代邻苯二胺、二酮类化合物和式(iv)所示的酯类化合物作为原料,通过亲核加成反应和碳碳交叉偶联反应制得所述的多环稠合的1,5
‑
苯并二氮杂卓化合物;
[0014][0015]
其中,r5为乙炔基、
[0016]
制备式(i)所示的化合物时,所述二酮类化合物的结构如式(v)所示:
[0017][0018]
制备式(ⅱ)所示的化合物时,所述二酮类化合物的结构如式(vi)所示:
[0019][0020]
其中,n为1或2。
[0021]
目前已经有很多关于1,5
‑
苯并二氮杂卓化合物合成方法的报道,传统的合成1,5
‑
苯并二氮杂卓化合物的方法大多是利用邻苯二胺与α,β
‑
不饱和羰基化合物或1,3
‑
二羰基化合物(或酮)的缩合反应;或是通过在分子内构建碳氮双键和碳碳双键,形成烯胺
‑
亚胺结构的活性中间体合成1,5
‑
苯并二氮杂卓化合物。利用分子内烯胺
‑
烯胺自环合的方法合成1,5
‑
苯并二氮杂卓化合物还未见报道。本发明首次利用分子内烯胺
‑
烯胺自环合的合成方
法,合成了未见文献报道的既具有稠合结构又含有
‑
ch2coor的1,5
‑
苯并二氮杂卓类化合物,对于扩展1,5
‑
苯并二氮杂卓化合物在医药和工业生产领域的应用具有十分重要的价值。
[0022]
优选的,所述多环稠合的1,5
‑
苯并二氮杂卓化合物的制备方法,包括如下步骤:
[0023]
将所述取代邻苯二胺和二酮类化合物加入有机溶剂中,混合均匀,于50
‑
110℃反应2
‑
4h,加入所述酯类化合物和酸催化剂,混合均匀,于10
‑
30℃反应3
‑
8h,得多环稠合的1,5
‑
苯并二氮杂卓化合物,反应方程式如下:
[0024][0025]
上述反应中,当r5为乙炔基时,r3为h;当r5为时,r3为
‑
ch3;当r5为时,r3为
‑
ch2ch3。
[0026]
以酯类化合物为制备式(i)所示的化合物为例,本发明提供的多环稠合的1,5
‑
苯并二氮杂卓化合物的合成过程的反应机理如下:
[0027][0028]
取代邻苯二胺的一个氨基作为亲核试剂,进攻5,5
‑
二甲基环己烷
‑
1,3
‑
二酮中的一个羰基碳,发生亲核加成反应再除去一分子水后得到烯胺型的中间产物5a,中间产物5a
的另一个氨基再与乙酸乙酰乙酯中经三氯化铈活化的羰基发生亲核加成反应再消除一分子水,得到烯胺
‑
烯胺结构的活性中间体6aa,然后中间体6aa经分子内环化得到中间体7aa,中间体7aa经分子内质子转移得到目标产物4aa。
[0029]
本发明通过一锅法合成了一种新型结构的多环稠合的1,5
‑
苯并二氮杂卓化合物,减少了中间体分离纯化的步骤,步骤简单,操作方便,更加适合工业化大规模生产应用,且产率及纯度较高,产率可达80%以上,纯度可达95
‑
98%。
[0030]
优选的,所述取代邻苯二胺、二酮类化合物和酯类化合物的物质的量比为1:0.8
‑
1:1
‑
2。
[0031]
更优选的,所述取代邻苯二胺、二酮类化合物和酯类化合物的物质的量比为1:0.9:1.2。
[0032]
优选的反应物质的比可以保证在用量较小的条件下,促进反应正向进行,提高目标产物的产率。
[0033]
优选的,所述酸催化剂与所述取代邻苯二胺的物质的量比为0.1
‑
0.2:1。
[0034]
更优选的,所述酸催化剂与所述取代邻苯二胺的物质的量比为0.1:1。
[0035]
优选的,所述酸催化剂为三氯化铈、冰醋酸、扁桃酸或对甲苯磺酸中至少一种。
[0036]
更优选的,所述酸催化剂为三氯化铈。
[0037]
优选的酸性催化剂可以保证在催化剂用量最少的前提,最大限度的提高反应活性,促进碳碳偶联反应的进行,提高反应速率以及产物收率;用量过少,催化效果不明显;用量过多,产率提高不明显,且造成催化剂的浪费以及产物纯度的降低。
[0038]
优选的,所述有机溶剂为无水甲醇、无水乙醇、1,2
‑
二氯乙烷、乙腈或甲苯中至少一种。
[0039]
更优选的,所述有机溶剂为无水乙醇。
[0040]
选择无水乙醇作为反应溶剂,可以更好地促进亲核加成反应和分子内环合反应的进行,提高反应速率,减少副反应的发生,且相比其他有机溶剂,无水乙醇更加绿色环保。
[0041]
优选的,所述有机溶剂与所述取代邻苯二胺的物质的量比为2
‑
8:1。
[0042]
优选的有机溶剂的用量可以使取代邻苯二胺和二酮类化合物充分溶解,促进反应的进行,提高中间产物的收率,同时,还可以保证中间产物和酯类化合物反应时能够充分溶解,且可保证目标产物生成后容易析出,提高目标产物收率。
[0043]
优选的,取代邻苯二胺和二酮类化合物的反应生成中间体的温度为有机溶剂的回流温度。
[0044]
更优选的,当有机溶剂为无水乙醇时,取代邻苯二胺和二酮类化合物的反应温度为无水乙醇回流温度。
[0045]
中间产物与式(iv)所示的酯类化合物的反应为合成多环稠合的1,5
‑
苯并二氮杂卓化合物的关键步骤。
[0046]
优选的,中间产物与式(iv)所示的酯类化合物的反应为合成多环稠合的1,5
‑
苯并二氮杂卓化合物的反应温度为20
‑
25℃。
[0047]
若降低反应温度,需要更长的反应时间或反应不会发生;提高反应温度,会导致副反应的发生,目标产物的收率和纯度均会降低。优选的反应温度,可以在最大限度降低副反应的前提下,保证目标产物的收率。
[0048]
本发明提供了一条新颖、温和且绿色的一锅法制备1,5
‑
苯并二氮杂卓化合物的方法,且制备得到了结构上含有烷氧羰基甲基(
‑
ch2coor)多环稠合的化合物,其结构的引入有望有效提高药物的生物活性,为研究具有高生理活性的新型药物提供了基础,对于扩展1,5
‑
苯并二氮杂卓化合物在医药和工业生产领域的应用具有十分重要的价值,推广价值极高。
具体实施方式
[0049]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0050]
为了更好的说明本发明,下面通过实施例做进一步的举例说明。
[0051]
实施例1
[0052]2‑
(7,10
‑
二甲基
‑1‑
氧代
‑
1,2,3,4,9,10
‑
六氢苯并[b]环戊[e][1,4]二氮杂
‑
10
‑
基)乙酸乙酯的制备:
[0053]
取一50ml洁净反应瓶,加入1.22g 4
‑
甲基邻苯二胺,0.882g 1,3
‑
环戊二酮和3ml无水乙醇,搅拌溶解后,于回流条件下反应2.5h,冷却至室温,向反应瓶中加入1.56g乙酰乙酸乙酯和0.37g三氯化铈,于15℃继续反应4.0h,反应结束后,冰浴析出固体,真空抽滤,无水乙醇洗涤,得灰色固体281mg,产率90%,纯度98%,熔点为216
‑
218℃,反应方程式如下。
[0054][0055]
ir(kbr,cm
‑1):3321,3237,2978,1711,1618,1561。
[0056]1h nmr(400mhz,dmso
‑
d6,tms):δ1.14(3h,t,j=7.0hz,me),1.41(3h,s,me),2.16(3h,s,me),2.21(2h,m,ch2),2.57(2h,m,ch2),2.71(1h,d,j=14.3hz,1/2ch2),2.76(1h,d,j=14.2hz,1/2ch2),3.93(2h,q,j=7.1hz,ch2),4.92(1h,s,nh),6.62(1h,d,j=8.0hz,ph),6.75(1h,s,ph),6.87(1h,d,j=8.1hz,ph),9.44(1h,s,nh)。
[0057]
13
c nmr(100mhz,dmso
‑
d6,tms):δ14.44,20.79,26.91,33.82,44.40,57.84,59.99,116.41,119.60,122.05,123.58,130.30,132.05,136.85,165.75,171.06,200.14。
[0058]
esi
‑
ms:c
18
h
22
n2o
3 314,found 315(m+1)。
[0059]
元素分析(%):c
18
h
22
n2o3:c 68.77,h 7.05,n 8.91;found:c 68.79,h 7.04,n 8.91。
[0060]
实施例2
‑4[0061]
实施例2
‑
4中2
‑
(7,10
‑
二甲基
‑1‑
氧代
‑
1,2,3,4,9,10
‑
六氢苯并[b]环戊[e][1,4]二氮杂
‑
10
‑
基)乙酸乙酯的制备方法与实施例1相同,具体的反应条件参见表1。
[0062]
表1实施例2
‑
4反应条件
[0063][0064][0065]
实施例2
‑
4制备的产品的纯度均可达到95
‑
98%。
[0066]
实施例5
[0067]2‑
(2,2,11
‑
三甲基
‑1‑
氧代
‑
2,3,4,5,10,11
‑
六氢
‑
1h
‑
二苯并[b,e][1,4]二氮杂
‑
11
‑
基)乙酸乙酯的制备:
[0068]
取一50ml洁净反应瓶,加入1.08g邻苯二胺,1.26g 5,5
‑
二甲基环己烷
‑
1,3
‑
二酮和3ml无水乙醇,于回流条件(第一反应温度)下反应3h,冷却至室温,向反应瓶中加入1.56g乙酰乙酸乙酯和0.37g三氯化铈,于20℃(第二反应温度)继续反应5h,反应结束后,冰浴析出白色固体,真空抽滤,无水乙醇洗涤,得白色固体274mg,产率82%,纯度98%,熔点为176
‑
178℃,反应方程式如下。
[0069][0070]
ir(kbr,cm
‑1):3352,3307,2949,1725,1625,1539。
[0071]1h nmr(400mhz,dmso
‑
d6,tms):δ1.02(3h,s,me),1.05(3h,s,me),1.13(3h,t,j=7.1hz,me),1.44(3h,s,me),2.06(2h,s,ch2),2.47(2h,s,ch2),2.89(1h,d,j=14.7hz,1/2ch2),2.99(1h,d,j=14.6hz,1/2ch2),3.93(2h,q,j=6.8hz,ch2),4.86(1h,s,nh),6.80
‑
6.82(2h,m,ph),6.93
‑
6.95(1h,m,ph),6.98
‑
7.00(1h,m,ph),8.61(1h,s,nh)。
[0072]
13
c nmr(100mhz,dmso
‑
d6,tms):δ14.53,27.85,28.25,28.82,30.51,44.49,46.01,51.71,59.76,60.30,113.33,119.94,121.56,122.56,123.29,133.72,137.80,
153.25,171.38,194.43。
[0073]
esi
‑
ms:c
20
h
26
n2o
3 342,found 343(m+1)。
[0074]
元素分析(%):c
20
h
26
n2o3:c 70.15,h 7.65,n 8.18;found:c 70.16,h 7.63,n 8.19。
[0075]
实施例6
[0076]2‑
(7
‑
溴
‑1‑
氧代
‑
1,2,3,4,9,10
‑
六氢苯并[b]环戊[e][1,4]二氮杂
‑
10
‑
基)乙酸乙酯的制备:
[0077]
取一50ml洁净反应瓶,加入1.85g 4
‑
溴邻苯二胺,0.882g 1,3
‑
环戊二酮和3ml无水乙醇,搅拌溶解后,于回流条件下反应3h,冷却至室温,向反应瓶中加入1.176g丙炔酸乙酯和0.37g三氯化铈,于25℃继续反应5.2h,反应结束后析出固体,真空抽滤,无水乙醇洗涤,得灰色固体290mg,产率89%,纯度98%,熔点为260
‑
262℃,反应方程式如下。
[0078][0079]
ir(kbr,cm
‑1):3371,3261,2956,1711,1618,1539。
[0080]1h nmr(400mhz,dmso
‑
d6,tms):δ1.19(3h,t,j=7.1hz,me),2.17(1h,dd,j=14.2,9.6hz,1/2ch2),2.27(2h,m,ch2),2.31(1h,d,j=14.2,4.4hz,1/2ch2),2.62(2h,m,ch2),4.03(2h,q,j=7.2hz,ch2),4.28(1h,m,j=9.3hz,ch),5.83(1h,d,j=4.7hz,nh),6.93
‑
6.94(2h,m,ph),6.99(1h,s,ph),9.73(1h,s,nh)。
[0081]
13
c nmr(100mhz,dmso
‑
d6,tms):δ14.42,26.83,33.29,42.04,50.71,60.55,115.25,121.97,123.53,125.11,131.48,139.13,166.85,171.03,199.58。
[0082]
esi
‑
ms:c
16
h
17
brn2o
3 364,found 365(m+1)。
[0083]
元素分析(%):c
16
h
17
brn2o3:c 52.62,h 4.69,n 7.67;found:c 52.63,h 4.70,n 7.66。
[0084]
实施例7
[0085]2‑
(8
‑
氯
‑1‑
氧代
‑
2,3,4,5,10,11
‑
六氢
‑
1h
‑
二苯并[b,e][1,4]二氮杂
‑
11
‑
基)乙酸乙酯的制备:
[0086]
取一50ml洁净反应瓶,加入1.42g 4
‑
氯邻苯二胺,1.008g 1,3
‑
环己二酮和3ml无水乙醇,搅拌溶解后,于汇流条件下反应3.5h,冷却至室温,向反应瓶中加入1.166g丙炔酸乙酯和0.37g三氯化铈,于30℃继续反应5.3h,反应结束后,冰浴析出固体,真空抽滤,无水乙醇洗涤,得白色固体240mg,产率79%,纯度98%,熔点为194
‑
196℃,反应方程式如下。
[0087][0088]
ir(kbr,cm
‑1):3293,3231,2949,1725,1618,1525。
[0089]1h nmr((400mhz,dmso
‑
d6,tms):δ1.11(3h,t,j=7.1hz,me),1.86(2h,m,ch2),
2.10(1h,dd,j=16.1,11.1hz,1/2ch2),2.27(2h,m,ch2),2.35(1h,dd,j=16.2,4.0hz,1/2ch2),2.59(2h,m,ch2),4.00(2h,q,j=7.2hz,ch2),4.65(1h,m,j=8.6hz,ch),5.50(1h,d,j=6.4hz,nh),7.04(1h,s,ph),7.27(1h,d,j=8.8hz,ph),7.36(1h,d,j=13.4hz,ph),9.30(1h,s,nh)。
[0090]
13
c nmr(100mhz,dmso
‑
d6,tms):δ14.57,18.98,21.45,27.86,36.68,38.60,56.52,60.15,112.41,114.65,115.99,121.38,132.08,134.72,147.66,154.78,173.48,196.10。
[0091]
esi
‑
ms:c
17
h
19
cln2o
3 334,found 335(m+1)。
[0092]
元素分析(%):c
17
h
19
cln2o3:c 60.99,h 5.72,n 8.37;found:c 60.98,h 5.74,n 8.36。
[0093]
实施例8
[0094]2‑
(3,3,11
‑
三甲基
‑1‑
氧代
‑
2,3,4,5,10,11
‑
六氢
‑
1h
‑
二苯并[b,e][1,4]二氮杂
‑
11
‑
基)乙酸丙酯的制备:
[0095]
取一50ml洁净反应瓶,加入1.08g邻苯二胺,1.26g 5,5
‑
二甲基环己烷
‑
1,3
‑
二酮和3ml无水乙醇,搅拌溶解后,于汇流条件下反应3h,冷却至室温,向反应瓶中加入1.728g乙酰乙酸丙酯和0.37g三氯化铈,于30℃继续反应5.1h,反应结束后,冰浴析出固体,真空抽滤,无水乙醇洗涤,得白色固体238mg,产率83%,纯度98%,熔点为162
‑
164℃,反应方程式如下。
[0096][0097]
ir(kbr,cm
‑1):3365,3307,2964,1711,1611,1532。
[0098]1h nmr((400mhz,dmso
‑
d6,tms):(400mhz,dmso
‑
d6,tms):δ0.85(3h,t,j=7.4hz,me),1.02(3h,s,ch3),1.05(3h,s,ch3),1.45(3h,s,ch3),1.53(2h,m,j=7.0hz,ch2),2.06(2h,s,ch2),2.47(2h,s,ch2),2.92(1h,d,j=14.7hz,1/2ch2),3.01(1h,d,j=14.7hz,1/2ch2),3.85(2h,t,j=6.6hz,ch2),4.84(1h,s,nh),6.80(2h,m,ph),6.93(1h,m,ph),7.00(1h,m,ph),8.60(1h,s,nh)。
[0099]
13
c nmr(100mhz,dmso
‑
d6,tms):δ10.80,21.95,27.86,28.31,28.84,30.52,44.44,46.07,51.76,60.30,65.34,113.39,119.98,121.58,122.56,123.33,133.77,137.84,153.27,171.50,194.44。
[0100]
esi
‑
ms:c
21
h
28
n2o
3 356,found 357(m+1)。
[0101]
元素分析(%):c
21
h
28
n2o3:c 70.76,h 7.92,n 7.86;found:c 70.78,h 7.94,n 7.85。
[0102]
实施例9
[0103]2‑
(3,3,7,8,11
‑
五甲基
‑1‑
氧代
‑
2,3,4,5,10,11
‑
六氢
‑
1h
‑
二苯并[b,e][1,4]二氮杂
‑
11
‑
基)乙酸乙酯的制备:
[0104]
取一50ml洁净反应瓶,加入1.36g 4,5
‑
二甲基
‑
1,2
‑
苯二胺,1.26g 5,5
‑
二甲基环己烷
‑
1,3
‑
二酮和3ml无水乙醇,搅拌溶解后,于汇流条件下反应2.8h,冷却至室温,向反应
瓶中加入1.56g乙酰乙酸乙酯和0.37g三氯化铈,于30℃继续反应4.8h,反应结束后,冰浴析出固体,真空抽滤,无水乙醇洗涤,得白色固体278mg,产率86%,纯度98%,熔点为156
‑
158℃,反应方程式如下。
[0105][0106]
ir(kbr,cm
‑1):3293,3242,2956,1725,1625。
[0107]1h nmr((400mhz,dmso
‑
d6,tms):(400mhz,dmso
‑
d6,tms):δ1.01(3h,s,me),1.02(3h,s,me),1.14(3h,t,j=7.1hz,me),1.42(3h,s,me),2.03(2h,s,ch2),2.08(3h,s,me),2.08(3h,s,me),2.42(2h,s,ch2),2.83(1h,d,j=14.6hz,1/2ch2),2.95(1h,d,j=14.7hz,1/2ch2),3.95(2h,q,j=7.0hz,ch2),4.81(1h,s,nh),6.69(1h,s,ph),6.75(1h,s,ph),8.49(1h,s,nh)。
[0108]
13
c nmr(100mhz,dmso
‑
d6,tms):δ14.56,19.14,19.26,27.79,28.36,28.54,30.50,44.24,46.02,51.80,59.81,60.47,113.21,121.01,123.70,129.04,130.96,131.40,135.50,153.51,171.58,194.22。
[0109]
esi
‑
ms:c
22
h
30
n2o
3 370,found 371(m+1)。
[0110]
元素分析(%):c
22
h
30
n2o3:c 71.32,h 8.16,n 7.56;found:c 71.34,h 8.14,n 7.55。
[0111]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1