新型三环芳香杂环化合物,及其制备方法、药物组合物和应用与流程
1.本发明涉及小分子药物领域,具体地,本发明提供了一种可以用于治疗与pd
‑
1/pd
‑
l1信号通路有关的疾病的小分子化合物。
2.发明背景
3.免疫系统对控制及根治很多疾病起着至关重要的作用,像各种癌症、病毒引起的疾病等。但癌症细胞经常能通过一些途径躲避或抑制免疫系统,从而快速繁殖。其中一个方式就是改变免疫细胞上表达的激活和抑制分子。阻断抑制免疫检查点,像pd
‑
1,证明是一个非常有效的抑制癌细胞的方法。
4.pd
‑
1是程序性细胞死亡蛋白
‑
1,也称之为cd279。它主要在激活的t细胞和b细胞中表达,功能是抑制细胞的激活,这是免疫系统的一种正常的自稳机制,因为过度的t/b细胞激活会引起自身免疫病,所以pd
‑
1是我们人体的一道保护墙。pd
‑
1是由268个氨基酸组成的i型跨膜糖蛋白,它的结构主要包括外免疫球蛋白可变区、疏水跨膜区以及胞内区。胞内区包含两个磷酸化位点,分别位于免疫受体酷氨酸抑制基序和免疫受体酷氨酸转换基序,这也证明pd
‑
1能够反向调节t细胞受体介导的信号。pd
‑
1有两个配体,pd
‑
l1和pd
‑
l2,它们在表达方式上不同。pd
‑
l1在多种肿瘤细胞中均有上调表达,它与t细胞上的pd
‑
1结合,抑制t细胞增殖和活化,使t细胞处于失活状态,最终诱导免疫逃逸。
5.pd
‑
1/pd
‑
l1发挥着反向免疫调节作用。当pd
‑
1与pd
‑
l1结合后,可致使t细胞的免疫受体酷氨酸转换基序结构域的酪氨酸多磷酸化,磷酸化的酪氨酸可结合磷酸酶蛋白酪氨酸酶2和蛋白酪氨酸酶1。这样即可阻碍细胞外信号调节激酶的活化,还可阻断磷脂肌醇3
‑
激酶(pi3k)和丝氨酸
‑
苏氨蛋白激酶(akt)的激活,从而抑制t淋巴细胞增殖和相关细胞因子的分沁。pd
‑
1/pd
‑
l1信号抑制t细胞活化和增殖的同时,还可使细胞因子白细胞介素2、干扰素γ和il
‑
10的分泌。此外,pd
‑
1/pd
‑
l1信号对b细胞也有类似的免疫功能,当pd
‑
1与b细胞抗原受体结合后,pd
‑
1细胞质区与含有蛋白酪氨酸酶2结合位点的酪氨酸酶发生作用,从而阻碍b细胞的活化。
6.基于pd
‑
1/pd
‑
l1的免疫疗法是新一代备受关注的免疫疗法。最近几年,一系列令人惊喜的研究结果证实pd
‑
1/pd
‑
l1抑制剂对多种肿瘤具有强大的抗肿瘤活性。目前已经上市的pd
‑
1/pd
‑
l1抗体调节剂有bms的ninolumab、merck的lambrolizumab以及roche的atezolizumab。除此之外还有很多在研的pd
‑
1/pd
‑
l1抗体调节剂,包括cure tech的pidilizumab、gsk的amp
‑
224和astrazeneca的medi
‑
4736。
7.虽然肿瘤免疫治疗被认为是靶向治疗后癌症治疗的新一代革命。但是目前上市和在研的pd
‑
1单抗药有其自身的缺陷,包括只能注射给药,不能口服,在体内不稳定,易被蛋白酶分解,易产生免疫交叉反应,纯化比较困难且生产成本高等。所以pd
‑
1/pd
‑
l1相互作用的小分子调节剂是肿瘤免疫治疗的更好选择。
8.综上所述,本领域迫切需要开发新型pd
‑
1/pd
‑
l1相互作用的小分子调节剂。
技术实现要素:
9.本发明的目的是提供一种新型pd
‑
1/pd
‑
l1相互作用的小分子调节剂。
10.本发明的第一方面,提供了一种如下式i所示的化合物,或其光学异构体、水合物、溶剂合物,或其药学上可接受的盐:
[0011][0012]
其中,n为0、1、2或3;
[0013]
m、p、q和t各自独立地选自0、1、2、3或4;
[0014]
x1、x2、x3、x4、x5和x6各自独立地选自下组:n、o、s、so、so2、c(r)2、nr;
[0015]
y1、y2、y3、y4、y5和y6各自独立地选自下组:n、ch、c;
[0016]
选自下组:取代或未取代的c1
‑
c15芳基、或取代或未取代的具有1
‑
4个杂原子的4
‑
12元(优选5
‑
7元)杂芳基、取代或未取代的4
‑
12元杂环基、取代或未取代的4
‑
12元的c5
‑
c12环烷基;
[0017]
选自下组:取代或未取代的c1
‑
c15亚芳基、或取代或未取代的具有1
‑
4个杂原子的4
‑
12元(优选5
‑
7元)亚杂芳基、取代或未取代的4
‑
12元亚杂环基、取代或未取代的4
‑
12元的c5
‑
c12亚环烷基;
[0018]
r、r1、r2、r3、r4和r5各自独立地选自下组:h、
‑
cn、三氟甲基、磺酰氨基、硝基、羟基、卤素、
‑
s
‑
r8、
‑
s(o)
‑
r8、
‑
s(o)2‑
r8、取代或未取代的c1
‑
c10烷基、取代或未取代的c2
‑
c6烯基、取代或未取代的c2
‑
c6炔基、取代或未取代的c1
‑
c6烷氧基、取代或未取代的c3
‑
c8环烷基、氧代(即=o)、=nrf、
‑
cn、羟基、nrdre(例如氨基)、取代或未取代的c1
‑
c6胺基、取代或未取代的
‑
(c1
‑
c6亚烷基)
‑
nh
‑
(c1
‑
c6亚烷基)、羧基、取代或未取代的c6
‑
c10芳基、取代或未取代的具有1
‑
3个杂原子的5
‑
12元杂芳基、取代或未取代的具有1
‑
4个杂原子的3
‑
12元杂环基,取代或未取代的取代或未取代的或
‑
(l
1a
)
r
‑
(l
2a
)
s
‑
(l
3a
)
s
‑
;
‑
c0‑8‑
o
‑
r8,
‑
c0‑8‑
c(o)or8,
‑
c0‑8‑
oc(o)or8,
‑
c0‑8‑
nr8r9,
‑
c0‑8‑
n(r8)c(o)r9,
‑
c0‑8‑
c(o)nr8r9;
[0019]
或r1和r2与其相连的环原子共同组成5,6或7元碳环或5,6或7元杂环,其中,所述的杂环含有1、2或者3个选自n,s或o;所述的碳环或杂环可以为与b环共轭的芳香环,也可为含不饱和键的非芳香环,或饱和环;
[0020]
r8和r9各自独立地选自下组:h、羟基,取代或未取代的c1
‑
c10烷基、取代或未取代的c2
‑
c6烯基、取代或未取代的c2
‑
c6炔基、取代或未取代的c1
‑
c6烷氧基、取代或未取代的c3
‑
c8环烷基、氧代(即=o)、=nrf、
‑
cn、羟基、nrdre(例如氨基)、取代或未取代的c1
‑
c6胺
基、取代或未取代的
‑
(c1
‑
c6亚烷基)
‑
nh
‑
(c1
‑
c6亚烷基)、羧基、取代或未取代的c6
‑
c10芳基、取代或未取代的具有1
‑
3个杂原子的5
‑
12元杂芳基、取代或未取代的具有1
‑
4个杂原子的3
‑
12元杂环基,取代或未取代的取代或未取代的或
‑
(l
1a
)
r
‑
(l
2a
)
s
‑
(l
3a
)
s
‑
;
[0021]
各个l
1a
各自独立地为选自下组的基团;化学键、取代或未取代的c1‑
c7亚烷基、取代或未取代的c2
‑
c4亚烯基、取代或未取代的c2
‑
c4亚炔基、
‑
s
‑
、
‑
o
‑
、取代或未取代的
‑
nh
‑
、
‑
s(o)
‑
、或
‑
s(o)2‑
;
[0022]
l
2a
选自下组:取代或未取代的c6
‑
c12亚芳基、取代或未取代的具有1
‑
3个杂原子的5
‑
12元亚杂芳基、取代或未取代的c3
‑
c8亚环烷基、取代或未取代的具有1
‑
3个杂原子的5
‑
10元亚杂环基;
[0023]
l
3a
选自下组:取代或未取代的c1
‑
c10烷基、c1
‑
c10芳基、
‑
cn、羟基、氨基、羧基、
‑
co
‑
nh
‑
so2‑
r
g
、
‑
nh
‑
so2‑
r
g
、
‑
so2‑
nh
‑
co
‑
r
g
;
[0024]
r为1、2、3、4、5、6;
[0025]
s分别为0、1、2;
[0026]
rd、re和rg各自独立地选自下组:h、取代或未取代的c1‑
c6烷基、取代或未取代的c3‑
c
10
环烷基、取代或未取代的c6‑
c
10
芳基;或rd和re共同形成取代或未取代的具有1
‑
3个选自n、s和o的杂原子的5
‑
10元杂环基;
[0027]
所述的rf选自下组:h、取代或未取代的c1‑
c6烷基、取代或未取代的c6‑
c
10
芳基、取代或未取代的c6‑
c
10
杂芳基、氰基、
‑
c(=o)
‑
nrdre、
‑
c(=o)
‑
取代或未取代的c1‑
c6烷氧基、
‑
c(=o)
‑
取代或未取代的c1‑
c6烷基、
‑
c(=o)
‑
取代或未取代的c3‑
c
10
环烷基、
‑
c(=o)
‑
取代或未取代的c2‑
c6烯基、
‑
c(=o)
‑
取代或未取代的c2‑
c6炔基;
[0028]
除非特别说明,所述的“取代”是指被选自下组的一个或多个(例如2个、3个、4个等)取代基所取代:卤素:包括但不限于
‑
f、cl、br、
‑
ch2cl、
‑
chcl2、
‑
ccl3、
‑
ch2f、
‑
chf2、
‑
cf3、氧代、
‑
cn、羟基、氨基、c1
‑
c6烷胺基、羧基、
‑
nhac、未取代或被一个或多个选自下组的取代基取代的选自下组的基团:c1
‑
c6烷基、c1
‑
c6烷氧基、c6
‑
c10芳基、c3
‑
c8环烷基、卤代的c6
‑
c10芳基、具有1
‑
3个选自n、s和o的杂原子的5
‑
10元杂芳基、具有1
‑
3个选自n、s和o的杂原子的5
‑
10元杂环基;所述的取代基选自下组:卤素、羟基、羧基、氰基、c1
‑
c6烷氧基、c1
‑
c6烷胺基;
[0029]
上述各式中,任一所述杂原子选自下组:b、p、n、s和o。
[0030]
在另一优选例中,r4为其中,r
b
和r
c r
d
各自独立地选自下组:h、取代或未取代的c1‑
c8烷基;或所述的r
b
和r
c
与相邻的n原子共同构成取代或未取代的具有1
‑
3个选自n、s和o的杂原子的4
‑
10元杂环基。
[0031]
在另一优选例中,所述的为选自下组的环形成的二价基团:所述的为选自下组的环形成的二价基团:
[0032][0032]
其中,所述的环的成键位置可以为n或c。
[0033]
在另一优选例中,所述的r3选自下组:甲基、
‑
f、cl、br、
‑
ch2cl、
‑
chcl2、
‑
ccl3、
‑
ch2f、
‑
chf2、
‑
cf3、
‑
cn、羟基、氨基。
[0034]
在另一优选例中,所述的为选自下组的结构:
[0035][0036]
且所述的r1和r2各自独立地选自下组:h、卤素、取代或未取代的c1
‑
c10烷基。
[0037]
在另一优选例中,所述的为选自下组的结构:
[0038]
[0039][0040]
在另一优选例中,所述的为如下所示的结构:
[0041][0042]
在另一优选例中,所述的为选自下组的结构:
[0043][0044]
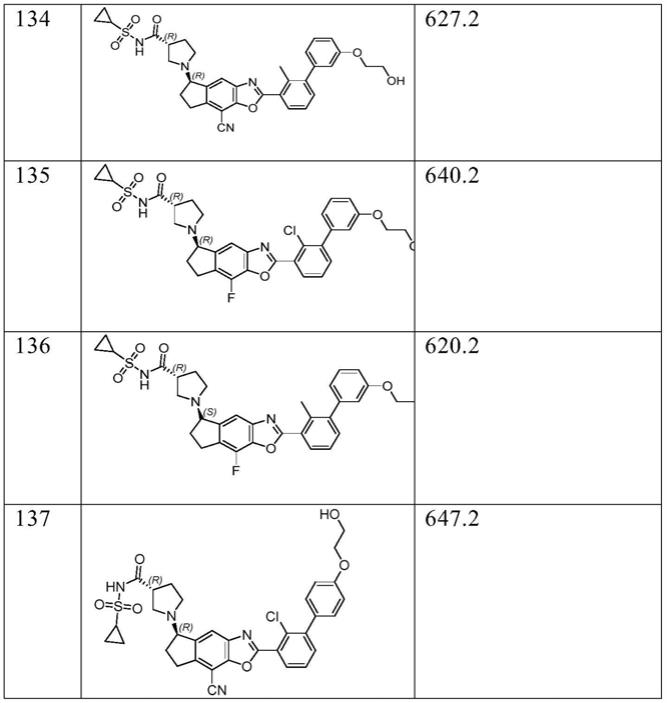
在另一优选例中,所述的化合物选自下列化合物;
[0045]
[0046]
[0047]
[0048]
[0049][0050]
本发明的第二方面,提供了一种如本发明第一方面所述的式i化合物的制备方法,其特征在于,所述的方法包括选自合成方案1或2所示的步骤:
[0051]
合成方案1:
[0052][0053]
(a)以中间体1和2为原料,通过钯催化剂催化的sonogashira偶联反应得到目标产物i
‑
1;
[0054]
优选地,中间体1的制备方法如下:
[0055][0056]
以化合物1
‑
1为原料,在钯催化剂和配体的条件下,发生偶联反应形成中间体1
‑
2;
[0057]
(b)以1
‑
2为原料,在路易斯酸催化下,与一级以及二级胺发生缩水反应,加入适当的还原剂,进一步还原形成中间体1
‑
3;或者在还原剂存在下直接还原胺化反应,形成中间体1
‑
3;
[0058]
(c)以1
‑
3为原料,在路易斯酸催化下,发生卤代反应,得到中间体1;
[0059]
合成方案2:
[0060][0061]
(a)以化合物3
‑
1为原料,与硝化剂反应(如浓硫酸/nano3,浓硫酸/发烟硝酸等),形成中间体3
‑
2;
[0062]
(b)以3
‑
2为原料,在还原条件下(pd
‑
c/h2;锌粉/氯化铵;铁粉/醋酸等),发生还原反应,形成中间体3
‑
3;
[0063]
(c)以3
‑
3和3
‑
4为原料,在缩水剂条件下,发生缩合反应,加入合适的氧化剂(如ddq等)发生氧化反应,得到中间体3
‑
5;
[0064]
(d)以3
‑
5和3
‑
6为原料,在催化剂和配体的条件下,发生偶联反应,形成中间体i
‑
2。
[0065]
本发明的第三方面,提供了一种药物组合物,其包含(1)如本发明第一方面所述的化合物,或其立体异构体或互变异构体,或其药学上可接受的盐、水合物或溶剂化物;(2)药学上可接受的载体。
[0066]
本发明的第四方面,提供了一种如本发明第一方面所述的化合物或其立体异构体或互变异构体,或其药学上可接受的盐、水合物或溶剂化物或如本发明第三方面所述的药物组合物的用途,其用于制备预防和/或治疗与pd
‑
1/pd
‑
l1的活性或表达量相关的疾病的药物组合物。
[0067]
本发明的第五方面,提供了一种pd
‑
1/pd
‑
l1调节剂,所述调节剂包含本发明第一方面所述的化合物、或其立体异构体或互变异构体、或其药学上可接受的盐、水合物或溶剂化物。
[0068]
在另一优选例中,所述的药物组合物被用于治疗选自下组的疾病:癌症、感染性疾病、自身免疫性疾病。
[0069]
在另一优选例中,所述的癌症选自下组:胰腺癌,膀胱癌,结肠直肠癌,乳腺癌,前列腺癌,肾癌,肝细胞癌,肺癌,卵巢癌,宫颈癌,胃癌,食道癌,黑色素瘤,神经内分泌癌,中枢神经系统癌,脑癌,骨癌,软组织肉瘤,非小细胞肺癌癌症,小细胞肺癌或结肠癌、皮肤癌、肺癌、泌尿系肿瘤、血液肿瘤、胶质瘤、消化系统肿瘤、生殖系统肿瘤、淋巴瘤、神经系统肿瘤、脑瘤、头颈癌。
[0070]
在另一优选例中,所述癌症选自下组:急性淋巴细胞白血病(all),急性髓性白血病(aml),慢性淋巴细胞白血病(cll),小淋巴细胞性淋巴瘤(sll),骨髓增生异常综合征(mds),骨髓增生性疾病(mpd),慢性粒细胞白血病(cml),多发性骨髓瘤(mm),非霍奇金淋巴瘤(nhl),套细胞淋巴瘤(mcl),滤泡性淋巴瘤,waldestrom巨球蛋白血症(wm),t细胞淋巴瘤,b细胞淋巴瘤或弥漫性大b细胞淋巴瘤(dlbcl)。
[0071]
在另一优选例中,所述的感染性疾病选自细菌感染、病毒感染。
[0072]
在另一优选例中,所述的自身免疫性疾病选自器官特异性自身免疫病、系统性自身免疫病。
[0073]
在另一优选例中,所述的器官特异性自身免疫病包括慢性淋巴细胞性甲状腺炎、甲状腺功能亢进、胰岛素依赖型糖尿病、重症肌无力、溃疡性结肠炎、恶性贫血伴慢性萎缩性胃炎、肺出血肾炎综合症、原发性胆汁性肝硬化、多发性脑脊髓硬化症、急性特发性多神经炎。
[0074]
在另一优选例中,所述的系统性自身免疫病包括类风湿关节炎、系统性红斑狼疮、系统性血管炎、硬皮病、天疱疮、皮肌炎、混合性结缔组织病、自身免疫性溶血性贫血。
[0075]
在另一优选例中,所述的药物组合物还用于改善慢性乙型肝炎(chb)患者t细胞功能。
[0076]
在另一优选例中,所述的抑制剂还包括至少一种选自下组的治疗剂:纳武单抗,派姆单抗,atezolizumab,或伊匹单抗。
[0077]
本发明的第六方面,提供了一种体内调节pd
‑
1/pd
‑
l1相互作用的方法,其包括步骤:将本发明第一方面所述的化合物、或其立体异构体或互变异构体、或其药学上可接受的盐、水合物或溶剂化物与pd
‑
l1蛋白接触。
具体实施方式
[0078]
本发明人经过广泛而深入的研究,发现了一类具有优异的调节效果的pd
‑
1/pd
‑
l1相互作用调节剂。在此基础上,发明人完成了本发明。
[0079]
定义
[0080]
如本文所用,术语“烷基”包括直链或支链的烷基。例如c1‑
c8烷基表示具有1
‑
8个碳原子的直链或支链的烷基,例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基等。
[0081]
如本文所用,术语“烯基”包括直链或支链的烯基。例如c2‑
c6烯基指具有2
‑
6个碳原子的直链或支链的烯基,例如乙烯基、烯丙基、1
‑
丙烯基、异丙烯基、1
‑
丁烯基、2
‑
丁烯基、或类似基团。
[0082]
如本文所用,术语“炔基”包括直链或支链的炔基。例如c2‑
c6炔基是指具有2
‑
6个碳原子的直链或支链的炔基,例如乙炔基、丙炔基、丁炔基、或类似基团。
[0083]
如本文所用,术语“c3‑
c
10
环烷基”指具有3
‑
10个碳原子的环烷基。其可以是单环,例如环丙基、环丁基、环戊基、环己基、或类似基团。也可以是双环形式,例如桥环或螺环形式。
[0084]
如本文所用,术语“c1‑
c8烷胺基”是指被c1‑
c8烷基所取代的胺基,可以是单取代或双取代的;例如,甲胺基、乙胺基、丙胺基、异丙胺基、丁胺基、异丁胺基、叔丁胺基、二甲胺基、二乙胺基、二丙胺基、二异丙胺基、二丁胺基、二异丁胺基、二叔丁胺基等。
[0085]
如本文所用,术语“c1‑
c8烷氧基”是指具有1
‑
8个碳原子的直链或支链的烷氧基;例如,甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、异丁氧基、叔丁氧基等。
[0086]
如本文所用,术语“具有1
‑
3个选自下组n、s和o的杂原子的3
‑
10元杂环烷基”是指具有3
‑
10个原子的且其中1
‑
3个原子为选自下组n、s和o的杂原子的饱和或部分饱和的环状基团。其可以是单环,也可以是双环形式,例如桥环或螺环形式。具体的实例可以为氧杂环丁烷、氮杂环丁烷、四氢
‑
2h
‑
吡喃基、哌啶基、四氢呋喃基、吗啉基和吡咯烷基等。
[0087]
如本文所用,术语“c6‑
c
10
芳基”是指具有6
‑
10个碳原子的芳基,例如,苯基或萘基等类似基团。
[0088]
如本文所用,术语“具有1
‑
3个选自下组n、s和o的杂原子的5
‑
10元杂芳基”指具有5
‑
10个原子的且其中1
‑
3个原子为选自下组n、s和o的杂原子的环状芳香基团。其可以是单环,也可以是稠环形式。具体的实例可以为吡啶基、哒嗪基、嘧啶基、吡嗪基、三嗪基、吡咯基、吡唑基、咪唑基、(1,2,3)
‑
三唑基以及(1,2,4)
‑
三唑基、四唑基、呋喃基、噻吩基、异恶唑基、噻唑基、恶唑基等。
[0089]
本发明所述的基团除非特别说明是“取代的或未取代的”,否则本发明的基团均可被选自下组的取代基所取代:卤素、腈基、硝基、羟基、氨基、c1‑
c6烷基
‑
胺基、c1‑
c6烷基、c2‑
c6烯基、c2‑
c6炔基、c1‑
c6烷氧基、卤代c1‑
c6烷基、卤代c2‑
c6烯基、卤代c2‑
c6炔基、卤代c1‑
c6烷氧基、烯丙基、苄基、c6‑
c
12
芳基、c1‑
c6烷氧基
‑
c1‑
c6烷基、c1‑
c6烷氧基
‑
羰基、苯氧羰基、c2‑
c6炔基
‑
羰基、c2‑
c6烯基
‑
羰基、c3‑
c6环烷基
‑
羰基、c1‑
c6烷基
‑
磺酰基等。
[0090]
如本文所用,“卤素”或“卤原子”指f、cl、br、和i。更佳地,卤素或卤原子选自f、cl和br。“卤代的”是指被选自f、cl、br、和i的原子所取代。
[0091]
除非特别说明,本发明所描述的结构式意在包括所有的同分异构形式(如对映异构,非对映异构和几何异构体(或构象异构体)):例如含有不对称中心的r、s构型,双键的(z)、(e)异构体等。因此,本发明化合物的单个立体化学异构体或其对映异构体、非对映异构体或几何异构体(或构象异构体)的混合物都属于本发明的范围。
[0092]
如本文所用,术语“互变异构体”表示具有不同能量的结构同分异构体可以超过低
能垒,从而互相转化。比如,质子互变异构体(即质子移变)包括通过质子迁移进行互变,如1h
‑
吲唑与2h
‑
吲唑。化合价互变异构体包括通过一些成键电子重组而进行互变。
[0093]
如本文所用,术语“溶剂合物”是指本发明化合物与溶剂分子配位形成特定比例的配合物。
[0094]
如本文所用,术语“水合物”是指本发明化合物与水进行配位形成的配合物。
[0095]
活性成分
[0096]
如本文所用,“本发明化合物”指式i所示的化合物,并且还包括及式i化合物的各种晶型形式、药学上可接受的盐、水合物或溶剂合物。
[0097]
优选的本发明化合物包括化合物1
‑
360(包括各化合物的各类r构型和/或s构型的立体异构体,和/或e
‑
/z
‑
的顺反异构体)。
[0098]
在另一优选例中,所述的药用盐包括与无机酸、有机酸、碱金属离子、碱土金属离子或能提供生理上可接受的阳离子的有机碱结合形成的盐以及铵盐。
[0099]
在另一优选例中,所述的无机酸选自盐酸、氢溴酸、磷酸或硫酸;所述的有机酸选自甲磺酸、对甲苯磺酸、三氟乙酸、枸杞酸、马来酸酒石酸、富马酸、柠檬酸或乳酸;所述的碱金属离子选自锂离子、钠离子、钾离子;所述的碱土金属离子选自钙离子、镁离子;所述的能提供生理上可接受的阳离子的有机碱选自甲胺、二甲胺、三甲胺、哌啶、吗啉或三(2
‑
羟乙基)胺。
[0100]
在本发明范围内的所有这些盐都可采用常规方法制备。在所述的通式i化合物及其溶剂化物和其盐的制备过程中,不同的结晶条件可能出现多晶或共晶。
[0101]
式i化合物的制备
[0102]
为了制备本发明通式i所述的化合物,依据通式i的结构,本发明制备通式i化合物可以通过以下合成路线获得。
[0103]
方程式1:
[0104][0105]
(a)以中间体1和2为原料,通过合适的钯催化剂催化的偶联反应得到目标产物i
‑
1;
[0106]
中间体1的制备方法举例如下:
[0107][0108]
(a)以化合物1
‑
1为原料(x为cl,br or i),在合适的钯催化剂和配体的条件下,发生偶联反应形成中间体1
‑
2;
[0109]
(b)以1
‑
2为原料,在合适的路易斯酸催化下,与一级以及二级胺发生缩水反应,加入适当的还原剂,进一步还原形成中间体1
‑
3;或者在合适的还原剂存在下直接还原胺化反应,形成中间体1
‑
3;
[0110]
(c)以1
‑
3为原料,在合适的路易斯酸催化下,发生卤代反应,得到中间体1;
[0111]
其中,中间体2的制备方法如下:
[0112][0113]
(a)以化合物2
‑
1和2
‑
2为原料,在合适的钯催化剂和配体的条件下,发生偶联反应(如suzuki,buchwald等),得到中间体2
‑
3;
[0114]
(b)以2
‑
2为原料,使用合适的试剂,移除硅基保护基,得到中间体2。
[0115]
另外,上述反应中的起始原料及中间体容易得到,各步反应可依据已报道的文献或对本领域熟练技术人员来说可以用有机合成中的常规方法很容易合成。通式i所述化合物可以溶剂化物或非溶剂化物的形式存在,利用不同的溶剂进行结晶可能得到不同的溶剂化物。
[0116]
方程式2:
[0117][0118]
(a)以化合物3
‑
1为原料,与合适的硝化剂反应(如浓硫酸/nano3,浓硫酸/发烟硝酸等),形成中间体3
‑
2;
[0119]
(b)以3
‑
2为原料,在合适的还原条件下(pd
‑
c/h2;锌粉/氯化铵;铁粉/醋酸等),发生还原反应,形成中间体3
‑
3;
[0120]
(c)以3
‑
3和3
‑
4为原料,在合适的缩水剂条件下,发生缩合反应,加入合适的氧化剂(如ddq等)发生氧化反应,得到中间体3
‑
5;
[0121]
(d)以3
‑
5和2
‑
1为原料,在合适的催化剂和配体的条件下,发生偶联反应,形成中间体i
‑
2;
[0122]
另外,上述反应中的起始原料及中间体容易得到,各步反应可依据已报道的文献或对本领域熟练技术人员来说可以用有机合成中的常规方法很容易合成。通式i所述化合物可以溶剂化物或非溶剂化物的形式存在,利用不同的溶剂进行结晶可能得到不同的溶剂化物。
[0123]
药物组合物和施用方法
[0124]
由于本发明化合物具有优异的pd
‑
1/pd
‑
l1相互作用的调节活性,因此本发明化合
物及其各种晶型,药学上可接受的无机或有机盐,水合物或溶剂合物,以及含有本发明化合物为主要活性成分的药物组合物可用于预防和/或治疗(稳定、减轻或治愈)pd
‑
1/pd
‑
l1相互作用相关疾病(例如,癌症、感染性疾病、自身免疫性疾病)。
[0125]
本发明的药物组合物包含安全有效量范围内的本发明化合物及药学上可以接受的赋形剂或载体。其中“安全有效量”指的是:化合物的量足以明显改善病情,而不至于产生严重的副作用。通常,药物组合物含有1
‑
2000mg本发明化合物/剂,更佳地,含有10
‑
200mg本发明化合物/剂。较佳地,所述的“一剂”为一个胶囊或药片。
[0126]“药学上可接受的载体”指的是:一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性。“相容性”在此指的是组合物中各组份能和本发明化合物以及它们之间相互掺和,而不明显降低化合物的药效。药学上可以接受的载体部分例子有纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等)、明胶、滑石、固体润滑剂(如硬脂酸、硬脂酸镁)、硫酸钙、植物油(如豆油、芝麻油、花生油、橄榄油等)、多元醇(如丙二醇、甘油、甘露醇、山梨醇等)、乳化剂(如吐温)、润湿剂(如十二烷基硫酸钠)、着色剂、调味剂、稳定剂、抗氧化剂、防腐剂、无热原水等。
[0127]
本发明化合物或药物组合物的施用方式没有特别限制,代表性的施用方式包括(但并不限于):口服、肠胃外(静脉内、肌肉内或皮下)。
[0128]
用于口服给药的固体剂型包括胶囊剂、片剂、丸剂、散剂和颗粒剂。在这些固体剂型中,活性化合物与至少一种常规惰性赋形剂(或载体)混合,如柠檬酸钠或磷酸二钙,或与下述成分混合:(a)填料或增容剂,例如,淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸;(b)粘合剂,例如,羟甲基纤维素、藻酸盐、明胶、聚乙烯基吡咯烷酮、蔗糖和阿拉伯胶;(c)保湿剂,例如,甘油;(d)崩解剂,例如,琼脂、碳酸钙、马铃薯淀粉或木薯淀粉、藻酸、某些复合硅酸盐、和碳酸钠;(e)缓溶剂,例如石蜡;(f)吸收加速剂,例如,季胺化合物;(g)润湿剂,例如鲸蜡醇和单硬脂酸甘油酯;(h)吸附剂,例如,高岭土;和(i)润滑剂,例如,滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、十二烷基硫酸钠,或其混合物。胶囊剂、片剂和丸剂中,剂型也可包含缓冲剂。
[0129]
固体剂型如片剂、糖丸、胶囊剂、丸剂和颗粒剂可采用包衣和壳材制备,如肠衣和其它本领域公知的材料。它们可包含不透明剂,并且,这种组合物中活性化合物或化合物的释放可以延迟的方式在消化道内的某一部分中释放。可采用的包埋组分的实例是聚合物质和蜡类物质。必要时,活性化合物也可与上述赋形剂中的一种或多种形成微胶囊形式。
[0130]
用于口服给药的液体剂型包括药学上可接受的乳液、溶液、悬浮液、糖浆或酊剂。除了活性化合物外,液体剂型可包含本领域中常规采用的惰性稀释剂,如水或其它溶剂,增溶剂和乳化剂,例知,乙醇、异丙醇、碳酸乙酯、乙酸乙酯、丙二醇、1,3
‑
丁二醇、二甲基甲酰胺以及油,特别是棉籽油、花生油、玉米胚油、橄榄油、蓖麻油和芝麻油或这些物质的混合物等。
[0131]
除了这些惰性稀释剂外,组合物也可包含助剂,如润湿剂、乳化剂和悬浮剂、甜味剂、矫味剂和香料。
[0132]
除了活性化合物外,悬浮液可包含悬浮剂,例如,乙氧基化异十八烷醇、聚氧乙烯山梨醇和脱水山梨醇酯、微晶纤维素、甲醇铝和琼脂或这些物质的混合物等。
[0133]
用于肠胃外注射的组合物可包含生理上可接受的无菌含水或无水溶液、分散液、
悬浮液或乳液,和用于重新溶解成无菌的可注射溶液或分散液的无菌粉末。适宜的含水和非水载体、稀释剂、溶剂或赋形剂包括水、乙醇、多元醇及其适宜的混合物。
[0134]
本发明化合物可以单独给药,或者与其他药学上可接受的化合物(例如其他抗癌制剂)联合给药。
[0135]
联合给药时,所述药物组合物还包括与一种或多种(2种,3种,4种,或更多种)其他药学上可接受的化合物。该其他药学上可接受的化合物中的一种或多种(2种,3种,4种,或更多种)可与本发明的化合物同时、分开或顺序地用于预防和/或治疗pd
‑
1/pd
‑
l1相互作用相关疾病。
[0136]
使用药物组合物时,是将安全有效量的本发明化合物适用于需要治疗的哺乳动物(如人),其中施用时剂量为药学上认为的有效给药剂量,对于60kg体重的人而言,日给药剂量通常为1~2000mg,优选20~500mg。当然,具体剂量还应考虑给药途径、病人健康状况等因素,这些都是熟练医师技能范围之内的。
[0137]
本发明的主要优点包括:
[0138]
(1)本发明化合物对pd
‑
1/pd
‑
l1相互作用具有很高的调节活性,与pd
‑
l1蛋白具有很强的结合能力,并具有解除pd
‑
l1抑制ifnγ的能力。
[0139]
(2)本发明的化合物具有更好的溶解性较好;对正常细胞的毒性非常低,因而可以在较大的剂量范围内应用于治疗对象。
[0140]
(3)相较于现有技术的化合物,本发明的化合物具有更好的溶解性,因此具有良好的成药性,相较于现有化合物而言,本发明化合物在体内实验之中表现出良好的生物利用度,除此之外,相较于现有化合物,本发明的化合物极易制成药学上可接受的盐,因而有助于进一步形成制剂。
[0141]
(4)体内药效研究表明,本发明化合物无论从肿瘤体积还是重量上,都可显著的抑制皮下肿瘤的生长,并可明显增加小鼠血液中、脾脏中各淋巴细胞数量。
[0142]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
[0143]
以下实施例中所用的实验材料和试剂如无特别说明均可从市售渠道获得。
[0144]
通用材料及测试方法:
[0145]
实施例中涉及到的仪器及原料说明如下:
[0146]
核磁共振氢谱是bruker av
‑
400(400mhz)核磁仪分析得到。
[0147]
化学位移以四甲基硅烷为内标来记录,以ppm为单位来表示(cdc13:δ7.26ppm)。记录的数据信息如下:化学位移及其裂分和偶合常数(s:单重峰;d:双重峰;t:三重峰;q:四重峰;br:宽峰;m:多重峰)。
[0148]
质谱数据除其他需要,都采用菲尼根高级lcq公司(finnigan lcq advantage)的液质联用仪进行分析,所有反应都在干燥氩气保护的无水无氧条件下进行操作。固体金属有机化合物在氩气保护干燥箱中进行储藏。
[0149]
四氢呋喃和乙醚是经过蒸馏得到,蒸馏时在其中加入金属钠和二苯甲酮。二氯甲烷,戊烷和己烷是用氢化钙来处理。
[0150]
本发明中涉及的特殊原料和中间体由天津长森药业有限公司等订制加工提供,其他所有化学试剂从上海化学试剂公司、阿尔得里奇公司(aldrich)、阿克罗公司(acros)等试剂供应商购买。如合成过程中反应所需的中间体或产物不够下一步试验,则重复多次合成至足够数量为止。
[0151]
本发明所涉及的原料和试剂除特殊说明外均可市售或订制加工购买得到。
[0152]
本发明中化合物可含有一个或多个不对称中心,因此该系列化合物可为消旋或者单一对映体形式。本发明所制备的化合物是纯度高于95%的杂环化合物,每个最终产物的结构表征分别由ms或/和氢谱核磁共振(1h nmr)分析确定。以下通过实施例说明本发明各类化合物和中间体的合成。
[0153]
实施例1化合物001的合成
[0154]
步骤1
‑
1:
[0155][0156]
取原料1
‑
1(100g,1.0eq),氢氧化钠(27g,1.0eq),加入到水(500ml),甲醇(2000ml)中,褐色略浑,降温至内浴
‑
25℃左右,一次性加入nis(151g,1.0eq),然后自然升温反应,固体增多,后又逐渐溶解至黑褐色清澈,反应2小时,点板以及lcms跟踪.加水1000ml,加2m盐酸(400ml),体系弱酸性,过滤,mtbe漂洗2次,得固体:161g,收率:87%。ms
‑
apci:275[m+h]
+
.
[0157]
步骤1
‑
2:
[0158][0159]
取dmf(1000ml,2bv)加入反应瓶中,氮气保护,加入原料1
‑
2(500g,1.0eq),氰化亚铜(172g,1.05eq),氮气置换,升温至内浴115℃,反应反应6小时,lcms跟踪;后处理:加thf稀释,过滤,滤液浓缩,室温下加入氢氧化钠水溶液(1.1eq,6bv),调ph=8~9;过滤,滤液用2m的硫酸调ph=7,加入硫酸亚铁,活性炭,室温搅拌1小时,过滤,滤液加2m的硫酸(400ml)调ph=2~3,固体过滤,异丙醇(200ml)打浆30分钟,过滤,烘干得211g棕色固体。ms
‑
apci:174[m+h]
+
.
[0160]
步骤1
‑
3:
[0161][0162]
将4.5克化合物1
‑
3溶于200毫升dcm(于500毫升单口烧瓶中),加入6.3克nis,氩气
保护。室温反应过夜。tlc点板1
‑
2反应完全。反应液加2m hcl 100ml,分液,干燥。旋干过柱(hep
‑
dcm 1:1,then dcm)纯化得到6g产物,淡黄色固体。ms
‑
apci:300[m+h]
+
.
[0163]
步骤1
‑
4:
[0164][0165]
在50毫升三口瓶中加入945毫克化合物1
‑
4,636毫克化合物1
‑
5,25毫克cui和25毫克pd(pph3)2cl2,氩气保护,加入20毫升dmf和1.2毫升dipea,55℃反应3小时。tlc点板原料消失。反应液加入到300毫升冰水中,乙酸乙酯300毫升萃取两次,2m naoh洗涤一次,干燥旋干。过柱(dcm
‑
meoh 10:1)纯化,得到120mg浅黄色固体。ms
‑
apci:364[m+h]
+
.
[0166]
步骤1
‑
5:
[0167][0168]
将215毫克化合物1
‑
6,248毫克氮杂环丁烷
‑3‑
羧酸甲酯盐酸盐,125毫克na(cn)bh3溶于10毫升thf中,氩气保护,加入167毫克三乙胺,70℃反应24小时。反应液加入到100毫升冰水中,ea 100毫升萃取两次,干燥。过柱(dcm
‑
meoh100,50:1,10:1,5:1)纯化,得到53mg浅黄色固体1
‑
7,ms
‑
apci:463[m+h]
+
.
[0169]
步骤1
‑
6:
[0170][0171]
将53毫克原料1
‑
7溶于5毫升meoh,加入2毫升2m naoh,26度反应5小时。反应液40度旋干,残留固体加入50毫升水,2m hcl调节ph值为7,虑出固体,水洗,thf洗,干燥,进一步制备纯化得黄色固体5毫克。ms
‑
apci:449[m+h]
+
[0172]
应用合成化合物001的方法,同样合成了下列表中的化合物:
[0173]
[0174]
[0175]
[0176]
[0177][0178]
实施例2化合物026的合成
[0179]
步骤2
‑
1:
[0180][0181]
取原料1
‑
3(110g,1.0eq)加入到醋酸(660ml,6bv)中,浑浊,升温至内浴40℃,缓慢滴加发烟硝酸(84g,2.1eq),浑浊,然后保温反应1小时左右,lcms跟踪,3.5%原料剩余,后处理:降温至10℃,搅拌30分钟,过滤,hep漂洗,晾干得固体117g,收率:84%,纯度88%,原料8%,加入thf(600ml)外浴50℃打浆1小时,室温过滤,得固体106g,收率:76%。ms
‑
apci:219[m+h]
+
.
[0182]
步骤2
‑
2:
[0183][0184]
取原料2
‑
4(20g,1.0eq),tfa(12g,1.1eq)加入thf(400ml,20bv)中,浑浊,加入5%的铂碳(10~20%),氢气置换,室温反应,lcms跟踪,(反应速度受多因素影响),后处理:过滤,3bvthf漂洗,直接投下一步。ms
‑
apci:189[m+h]
+
.
[0185]
步骤2
‑
3:
[0186][0187]
将2.02克化合物2
‑
3溶于50毫升dmac中,加入1.98克2
‑
4,氩气保护,室温反应3小时。加入3克ddq,室温反应过夜。反应液倒入150毫升水中,dcm(100毫升)萃取两次,干燥。旋干过柱(dcm
‑
meoh 100,50:1,30:1,20:1,10:1,5:1)纯化得到100毫克产物,淡黄色固体。ms
‑
apci:387[m+h]
+
。
[0188]
步骤2
‑
4:
[0189][0190]
将中间体2
‑
7(20.7g,3.3eq)溶于thf(143ml,10v/w),加入中间体2
‑
8(14.3g,1.0eq),加入nabh3cn(5.0eq,11.6g),置换氮气。70℃油浴搅拌过夜。lc
‑
ms检测,2
‑
7剩余约15%,停止反应,降至室温,与另一批次1g反应合并,加水,ea萃取两次,合并有机相,食盐水洗,干燥浓缩。拌样过柱,meoh in dcm(2%)得产品17.2g及交叉点5.0g类白色固体。分别使用dcm:hep 1:1 100ml、20ml打浆,室温搅拌10分钟,过滤,hep淋洗,收集滤饼,分别干燥得10.3g、1.0g,lcms检测无明显杂质,合并得类白色固体11.1g,hplc 97.9%,收率51.8%。送手性拆分,得peak 1 4.98g,peak 2 5.12g。
[0191]
步骤2
‑
5:
[0192][0193]
将叔丁酯2
‑
9(1g,1.0eq)溶于tfa(10v/w),室温下搅拌1小时。lcms检测反应完全,直接浓缩,得黄色至棕黄色的油状物。向浓缩残留物中加入水(10v/w),有粘稠半油半固物质析出,用饱和nahco3溶液调ph至6
‑
7,有大量具黏性的半油半固物质聚集成团
(1)
。粗过滤一遍,收集滤饼(或成糊状),加水(20v/w),超声5~10分钟,粘稠物逐渐固化,并部分分散。
室温搅拌2小时,基本分散,过滤,水洗,收集滤饼,干燥得产品500mg,ms
‑
apci:486[m+h]
+
。
[0194]
步骤2
‑
6:
[0195][0196]
将化合物硼酸酯原料2
‑
11(commercial,37mg),2
‑
10(100mg),磷酸钾(88mg)和ruphos
‑
pd
‑
g3(5mg)依次加入装有1,4
‑
dioxane/h2o(4:1,5ml)的单口瓶中,氮气保护,于油浴90度搅拌1小时,lc
‑
ms检测反应。反应完全后,过滤,旋干溶剂,prep
‑
hplc制备得10mg化合物026(白色固体),esi(apci):542[m+h]
+
.
[0197]
1h nmr(400mhz,dmso
‑
d6)δ12.94(s,1h),10.34(s,1h),8.47(s,1h),8.08(dd,j=6.9,2.6hz,1h),7.77
–
7.39(m,2h),7.10
–
6.64(m,3h),5.12(s,1h),4.28(s,4h),3.67
–
3.4(m,3h),3.24
–
3.15(m,3h),2.37
–
2.10(m,2h),2.00
–
1.93(m,1h).
[0198]
应用合成化合物002的方法,合成了下列表中的化合物:
[0199]
[0200]
[0201]
[0202]
[0203]
[0204]
[0205]
[0206][0207]
实施例3化合物025的合成步骤3
‑
1:
[0208][0209]
将化合物3
‑
1(1125mg,2.32mmol),环丙基磺酰胺(310mg,2.56mmol),dmap(567mg,
4.64mmol)和edci(889mg,4.64mmol)依次加入装有dcm(15ml)的单口瓶中,室温搅拌1小时,tlc检测反应(紫外较弱,用0.5m稀盐酸洗涤之后点板)。反应完全后,用0.5m稀盐酸洗涤和卤水洗涤,无水硫酸钠干燥,柱层析,得白色固体548mg。esi(apci):589[m
‑
h]
‑
.
[0210]
步骤3
‑
2:
[0211][0212]
将化合物硼酸酯原料2
‑
11(commercial,28mg),2
‑
10(90mg),磷酸钾(88mg)和xantphos
‑
pdcl2(13mg)依次加入装有1,4
‑
dioxane/h2o(4:1,5ml)的单口瓶中,氮气保护,于油浴90度搅拌2小时,lc
‑
ms检测反应。反应完全后,过滤,旋干溶剂,prep
‑
hplc制备得10mg化合物025(白色固体),esi(apci):645[m+h]
+
.
[0213]
1h nmr(400mhz,dmso
‑
d6)δ8.50(s,1h),8.11(dd,j=6.8,2.6hz,1h),7.72
–
7.61(m,2h),6.99(dd,j=5.2,3.1hz,2h),6.94(dd,j=8.3,2.1hz,1h),5.17(d,j=7.0hz,1h),4.31(s,4h),3.49(d,j=8.2hz,2h),3.32(s,2h),3.24(ddd,j=17.2,8.6,3.1hz,2h),2.96(dq,j=12.8,6.6,5.9hz,2h),2.69
–
2.54(m,2h).
[0214]
[0215]
[0216][0217]
生物测试
[0218]
实施例a:pd
‑
1/pd
‑
l1均相时间分辨荧光(htrf)结合测定
[0219]
测定在标准黑色384孔聚苯乙烯板中进行,终体积为20μl。首先将抑制剂用dmso连续稀释后加入板孔中,再加入其他反应组分。测定中dmso的最终浓度为1%。测定是在25℃下含有0.05%tween
‑
20和0.1%bsa的pbs缓冲液(ph7.4)中进行的。在c端带有his标记的重组人pd
‑
l1蛋白(19
‑
238)购自acrobiosystems公司(pd1
‑
h5229)。在c端带有fc标记的重组人pd
‑
1蛋白(25
‑
167)也购自acrobiosystems公司(pd1
‑
h5257)。将pd
‑
l1和pd
‑
1蛋白在测定缓冲液中稀释然后提取0.1μl溶液加入到板孔中。离心平板并将蛋白质与抑制剂预孵育40分钟。孵育后加入0.1μl htrf检测缓冲液含有铕封闭标记的抗人igg(perkinelmer
‑
ad0212)fc专属的和抗his的
‑
别藻蓝蛋白(apc,perkinelmer
‑
ad0059h)缀合的抗体。离心后,将孔板在25℃下孵育60分钟。置于pherastar fs读板器中读取数据(665nm/620nm比率)。测定中的最终浓度为~3nm pd1、10nm pd
‑
l1、1nm铕抗人igg和20nm抗
‑
his
‑
别藻蓝蛋白。使用graphpad prism5.0软件拟合活性数据得出抑制剂的ic
50
值。
[0220]
实施例中举例说明的化合物ic
50
值以下列方式表示:ic
50
:+:≤100nm;++:100nm~1000nm;+++:>1000nm。
[0221]
使用实施例a中描述的pd
‑
1/pd
‑
l1均相时间分辨荧光(htrf)结合测定获得的实施例化合物的数据提供于表1中。
[0222]
[0223][0224]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1