一种富含硼酸酯和羟基的氟聚合物的合成方法
本发明属于聚合物合成技术领域,具体涉及一种富含硼酸酯和羟基的氟聚合物的合成方法。
背景技术:
由于氟原子的高电负性和碳-氟键的强离解能,含氟聚合物表现出许多独特的优异性能,如高的热稳定性和耐化学性(对溶剂、酸和碱)、低的折射率、低的表面能和介电常数。官能化的含氟聚合物结合了氟碳化合物的固有优点和功能取代基的物理化学性质,在离子交换/传导、涂层材料、自愈合材料等领域有着广泛的应用。例如,在聚四氟乙烯的侧链引入亲水的磺酸基团,得到全氟磺酸聚合物(pfsa)。该聚合物既具有憎水的聚四氟乙烯骨架,又含有亲水的磺酸基侧链,这种独特的结构赋予其出色的力学性能、优异的稳定性及较高的质子传导率,使其成功应用于燃料电池、储能电池和水处理等多种领域。杜邦公司通过将2,2-双(三氟甲基)-4,5-二氟-1,3-二氧杂环戊烯与四氟乙烯共聚,得到一种全氟无定形聚合物teflonaf,这种无定形聚合物具有高光学透明度和极低的折射指数,可用作光学镜头和防护涂层。然而,强的碳-氟键限制了含氟聚合物通过取代氟原子进行后修饰改性,直接实现含氟聚合物的官能化比较困难,因此,开发具有官能化的含氟聚合物的合成方法至关重要。
在官能化的含氟聚合物中,羟基官能化的氟聚合物是特殊的一类,如偕三氟甲基/羟基氟聚合物在光刻胶领域具有重要的应用(专利号:us20060036005a1)。目前在半导体光刻技术中常用的波长是365nm和248nm,但许多有机官能团在157nm处的高吸光度,使得很难开发出既可溶于碱性显影液,又在波长157nm处具有低吸光度的有机聚合物,从而阻碍了光刻技术的发展。偕三氟甲基/羟基氟聚合物既具有含氟聚合物的低吸光度特性,又含有酸性的羟基官能团,使其能够很好的应用于157nm的光刻胶中(专利号:wo2001063362a2)。目前偕三氟甲基/羟基氟聚合物只能通过2-(三氟甲基)醋酸乙烯酯与醋酸乙烯酯共聚物的水解得到(期刊号:journalofphotopolymerscienceandtechnology,2000,13,451-458),由于2-(三氟甲基)醋酸乙烯酯与醋酸乙烯酯具有较强的交替聚合倾向,共聚物中2-(三氟甲基)醋酸乙烯酯的摩尔分数不超过0.5(期刊号:macromolecules,2012,45,9674-9681),因此开发新的合成方法,合成出较高摩尔分数的偕三氟甲基/羟基氟聚合物,具有重要的价值。
在过去的几十年中,人们对一些含2-(三氟甲基)的烯基单体进行了聚合研究,如2-(三氟甲基)苯乙烯(专利号:us20100203450a1)、2-(三氟甲基)丙烯酸酯(专利号:cn104725544a)。这些三氟甲基取代的单体的一个主要特点是难以进行后修饰,只能得到特定结构的聚合物。研究开发一种新型可后修饰的2-(三氟甲基)的烯基单体,是合成偕三氟甲基/羟基氟聚合物的有效途径。最近,jäkle(期刊号:chem.commun.,2016,52,13616-13619)、klausen(专利号:wo2019079223a1)和ouchi(期刊号:angew.chem.int.ed.,2019,58,12435-12439;acsmacroletters,2020,9,788-793)分别研究了乙烯基硼烷单体的自由基聚合,提供了主链悬挂有硼烷聚合物的合成方法,这些主链悬挂的硼烷可以被羟基以及其他官能团取代,实现聚合物的后修饰。除了化学转化外,以硼酸酯为侧基的聚合物也因其在动态交联网络(期刊号:macromolecules,2020,53,956-964;adv.mater.,2016,28,8277-8282)、聚合物纳米结构(期刊号:polym.chem.,2011,2,2122-2132)等领域的应用而引起了人们的高度关注。
技术实现要素:
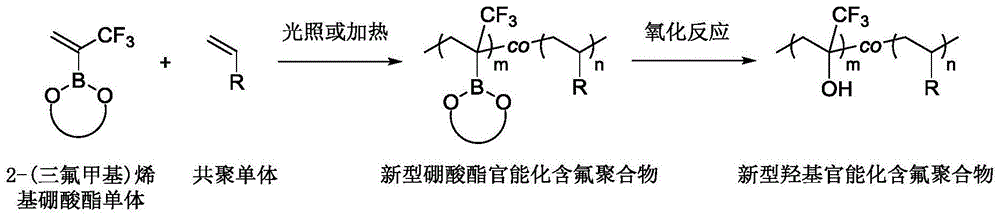
基于上述现有技术,本发明的目的在于提供一种富含硼酸酯和羟基的氟聚合物的合成方法;本发明通过2-(三氟甲基)烯基硼酸酯与烯基醚、烯基酯和烯基酰胺及其衍生物的共聚,通过调节共聚单体的投料比,得到了一系列不同组分比例的新型硼酸酯官能化含氟聚合物,改变调聚剂的添加量,可以调控含氟聚合物的分子量。此外,通过对聚合物进行后修饰,可以得到较高摩尔分数的偕三氟甲基/羟基氟聚合物,为新型富含硼酸酯和羟基的氟聚合物的合成提供了一种便捷易行的途径。本发明合成方法具有操作简单、适用单体范围广、聚合反应速率快等优点。
本发明的技术方案具体介绍如下。
一种富含硼酸酯和羟基的氟聚合物的合成方法,具体步骤如下:
步骤一、在无氧条件下,以2-(三氟甲基)烯基硼酸酯类化合物为第一单体,以乙烯基醚、乙烯基酯或乙烯基酰胺类化合物中的一种为第二单体,在光照或者加热条件下,分别在光引发剂或热引发剂作用下在反应溶剂中发生自由基聚合反应制备富含硼酸酯的氟聚合物;
步骤二、将富含硼酸酯的氟聚合物通过氧化反应进行后修饰,将硼酸酯全部或部分转化为羟基,得到富含羟基的氟聚合物。
本发明中,步骤一中,2-(三氟甲基)烯基硼酸酯类化合物结构如式(1)或式(2)所示:
其中:
r1、r2、r3、r4独立地选自氢原子、碳原子数为1~6的烷基或芳基中的任意一种;r5为单取代或多取代的氢原子、卤原子、碳原子数为1~6酯基或烷基。
本发明中,步骤一中,乙烯基醚类化合物的结构如式(3)所示,乙烯基酯类化合物的结构如式(4)所示,乙烯基酰胺类化合物的结构如式(5)或(6)所示:
其中:
r6选自碳原子数为1~12的烷基、碳原子数为5~12的环烷基、碳原子数为1~6的氯烷基、碳原子数为1~6的碘烷基、碳原子数为1~6的羟基烷基、重复单元为1~12的乙二醇基或碳原子数为1~6羧基烷基中的任意一种;r7为碳原子数为1~12的烷基或芳基;r8和r9独立地选自氢原子或碳原子数为1~12的烷基、芳基中的任意一种;n为1~6之间的整数。
本发明中,步骤一中,按照摩尔数计,第一单体与第二单体的投料比为第一单体:第二单体=10:(1~100)。
本发明中,步骤一中,光照条件下,使用发射波长为390~700nm的光源,反应时间为1~24小时。
本发明中,步骤一中,加热条件下,加热反应温度为30~120℃,加热反应时间为1~24小时。
本发明中,步骤一中,光引发剂为2-羟基-2-甲基-1-苯基丙酮、2,4,6-三甲基苯甲酰基-二苯基氧化膦或苯甲酰甲酸甲酯中的一种或几种;热引发剂为偶氮二异丁腈、偶氮二异庚腈、偶氮二异丁酸二甲酯或过氧化苯甲酰中的一种或几种;按照摩尔数计,(第一单体与第二单体的加和):光引发剂或热引发剂=1000:(1~100)。
本发明中,步骤一中,自由基聚合反应中,还加入调聚剂,调聚剂为二硫醚为骨架的化合物,调聚剂为第一单体和第二单体总量的0.0005~10mol%。优选的,调聚剂结构如式(7)所示:
其中:
r10、r11独立地选自碳原子数为1~12的烷基或芳基中任一种。
本发明中,步骤一中,反应溶剂为四氢呋喃、乙醚、二甲基亚砜,n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、乙腈、甲苯、碳酸二乙酯中的一种或多种。
本发明中,步骤二中,将富含硼酸酯的氟聚合物中的硼酸酯全部或部分转化为羟基的氧化反应条件如下:碱性条件下,双氧水作为氧化剂。
和现有技术相比,本发明的有益效果在于:
实验结果表明本发明方法通过加热或光照的自由基聚合方法,成功地进行了2-(三氟甲基)烯基硼酸酯与烯基醚、烯基酯和烯基酰胺及其衍生物的共聚,通过调节共聚单体的投料比,得到了一系列不同组分比例的新型硼酸酯官能化含氟聚合物,改变调聚剂的添加量,可以调控含氟聚合物的分子量,通过硼酸酯的后修饰,得到新型羟基官能化含氟聚合物,为新型富含硼酸酯和羟基的氟聚合物的合成提供了一种便捷易行的途径,本发明方法具有操作简单、适用单体范围广、无金属、聚合反应速率快等优点。
附图说明
图1为一种新型富含硼酸酯和羟基的氟聚合物的制备原理图。
图2为实施例1的(a)1hnmr和(b)19fnmr谱图。
图3为实施例2的(a)1hnmr和(b)19fnmr谱图。
图4为实施例4的(a)1hnmr和(b)19fnmr谱图。
图5为实施例10的(a)1hnmr和(b)19fnmr谱图。
图6为实施例11的19fnmr谱图。
图7为实施例3的gpc谱图。
具体实施方式
实施例中,提供一种富含硼酸酯和羟基的氟聚合物的合成方法,具体步骤如下:
(1)制备反应溶液:将调聚剂、引发剂、单体、溶剂分别加入到反应瓶中,搅拌混合均匀,并除去反应体系中的氧气;该反应溶液分为四种:
反应溶液a,含有第一单体、第二单体、热引发剂、溶剂;
反应溶液b,含有第一单体、第二单体、热引发剂、溶剂、调聚剂;
反应溶液c,含有第一单体、第二单体、光引发剂、溶剂;
反应溶液d,含有第一单体、第二单体、光引发剂、溶剂、调聚剂;
按摩尔比计,单体:引发剂=1000:(1~100);调聚剂为单体的0.0005~10mol%
(2)将反应溶液置于容器中,在搅拌条件下,对于反应溶液a和反应溶液b,加热反应时间为1~24小时,加热反应温度为30~120℃;对于反应溶液c和反应溶液d,在光照下反应时间为1~24小时,光照反应使用发射波长为390~700nm的光源。
(3)反应结束后,将反应溶液析出、纯化,即得到富含硼酸酯的氟聚合物;
(4)将硼酸酯官能化的氟聚合物通过氧化反应进行后修饰,再经过分离与纯化,即得到富含羟基的氟聚合物;
其中,所用第一单体为2-(三氟甲基)烯基硼酸酯;第二单体为乙烯基醚、乙烯基酯和乙烯基酰胺及其衍生物的一种或多种。
下面结合部分具体实施方案对本发明进行详述,这些实施例仅用于说明本发明,而不用于限制本发明的范围,实施例中的制备方案仅为优选方案,但本发明并不局限于优选制备方案。对于同一个反应,调整反应时间或反应装置可以在不改变反应条件参数的情况下实现不同分子量以及分子量分布的聚合物的合成。
第一部分:合成单体2-(三氟甲基)乙烯基硼酸频哪醇酯
实施例1:将带有搅拌子的三颈圆底烧瓶进行氮气置换三次,进行氮气保护,然后将50mmol的2-溴-3,3,3-三氟丙烯和100ml四氢呋喃加入到三颈圆底烧瓶中,在-78℃条件下缓慢滴加47ml浓度为1.3mol/l含有氯化锂的异丙基氯化镁溶液,待溶液滴加完毕,搅拌反应5个小时,再缓慢滴加60mmol三甲基硼酸酯,关闭制冷,继续搅拌反应过夜,使其缓慢恢复至室温。反应停止后,加入1mol/l盐酸溶液进行酸化,再加入乙醚进行萃取,合并有机相,然后向有机相中加入5g无水硫酸镁和60mmol频哪醇继续搅拌6个小时,过滤除去硫酸镁固体,得到粗产品,将粗产品进行柱层析分离,得到无色透明液体产物。产物的1hnmr如图2(a)所示,19fnmr谱图如图2(b)所示。
第二部分:合成新型硼酸酯官能化氟聚合物
实施例2:采用反应溶液a,通过加热合成2-(三氟甲基)乙烯基硼酸频哪醇酯与乙烯基正丁醚共聚物
按2-(三氟甲基)乙烯基硼酸频哪醇酯:乙烯基正丁醚:偶氮二异丁腈=100:100:1的摩尔比将上述原料50mmol、50mmol、0.5mmol分别加入到装有磁子的瓶子中,加入50mln,n-二甲基甲酰胺,搅拌溶解均匀后将反应混合物脱除氧气,随后使反应体系在65℃条件下反应24个小时,通过核磁共振氢谱测得2-(三氟甲基)乙烯基硼酸频哪醇酯转化率为98%,乙烯基正丁醚转化率为65%,聚合物样品用体积比为1:1的甲醇和水混合溶液沉淀三次,真空干燥至恒重,得到白色固体,聚合物的1hnmr谱图、19fnmr谱图分别如图3(a)、(b)所示。通过凝胶渗透色谱测得的聚合物分子量mn=8.1×103g/mol和分子量分布mw/mn=1.49。
实施例3:采用反应溶液b,通过加热合成2-(三氟甲基)乙烯基硼酸频哪醇酯与乙烯基正丁醚共聚物
按2-(三氟甲基)乙烯基硼酸频哪醇酯:乙烯基正丁醚:偶氮二异丁腈:对甲苯二硫醚=100:100:1:1的摩尔比将上述原料50mmol、50mmol、0.5mmol、0.5mmol分别加入到装有磁子的瓶子中,加入50mln,n-二甲基甲酰胺,搅拌溶解均匀后将反应混合物脱除氧气,随后使反应体系在65℃条件下反应24个小时,通过核磁共振氢谱测得2-(三氟甲基)乙烯基硼酸频哪醇酯转化率为98%,乙烯基正丁醚转化率为67%,聚合物样品用体积比为1:1的甲醇和水混合溶液沉淀三次,真空干燥至恒重,得到白色固体,聚合物的gpc谱图如图7所示。通过凝胶渗透色谱测得的聚合物分子量mn=6.7×103g/mol和分子量分布mw/mn=1.49。
实施例4:采用反应溶液c,通过光照合成2-(三氟甲基)乙烯基硼酸频哪醇酯与醋酸乙烯酯共聚物
按2-(三氟甲基)乙烯基硼酸频哪醇酯:醋酸乙烯酯:2,4,6-三甲基苯甲酰基-二苯基氧化膦=100:100:1的摩尔比将上述原料50mmol、50mmol、0.5mmol分别加入到装有磁子的瓶子中,加入50mln,n-二甲基甲酰胺,搅拌溶解均匀后将反应混合物脱除氧气,随后将反应体系放置在13w波长为400-750nm白色led灯照射下反应24个小时,通过核磁共振氢谱测得2-(三氟甲基)乙烯基硼酸频哪醇酯转化率为98%,醋酸乙烯酯转化率为86%,聚合物样品用体积比为1:1的甲醇和水混合溶液沉淀三次,真空干燥至恒重,得到白色固体,聚合物的1hnmr谱图、19fnmr谱图分别如图4(a)、(b)所示;通过聚合物的1hnmr谱图分析得到,相比于醋酸乙烯酯结构单元,偕三氟甲基/硼酸频哪醇酯结构单元在聚合物中的摩尔分数为0.47。通过凝胶渗透色谱测得的聚合物分子量mn=6.4×103g/mol和分子量分布mw/mn=1.67。
实施例5:采用反应溶液d,通过光照合成2-(三氟甲基)乙烯基硼酸频哪醇酯与n-乙烯基吡咯烷酮共聚物
按2-(三氟甲基)乙烯基硼酸频哪醇酯:n-乙烯基吡咯烷酮:2,4,6-三甲基苯甲酰基-二苯基氧化膦:对甲苯二硫醚=100:100:1:1的摩尔比将上述原料50mmol、50mmol、0.5mmol、0.5mmol分别加入到装有磁子的瓶子中,加入50mln,n-二甲基甲酰胺,搅拌溶解均匀后将反应混合物脱除氧气,随后将反应体系放置在13w波长为400-750nm白色led灯照射下反应24个小时,通过核磁共振氢谱测得2-(三氟甲基)乙烯基硼酸频哪醇酯转化率为98%,n-乙烯基吡咯烷酮96%,聚合物样品用体积比为1:1的甲醇和水混合溶液沉淀三次,真空干燥至恒重,得到白色固体。通过凝胶渗透色谱测得的聚合物分子量mn=1.1×104g/mol和分子量分布mw/mn=1.50。
实施例6:采用反应溶液d,通过光照合成较高分子量的2-(三氟甲基)乙烯基硼酸频哪醇酯与乙烯基正丁醚共聚物
按2-(三氟甲基)乙烯基硼酸频哪醇酯:乙烯基正丁醚:2,4,6-三甲基苯甲酰基-二苯基氧化膦:对甲苯二硫醚=200:200:1:1的摩尔比将上述原料50mmol、50mmol、0.25mmol、0.25mmol分别加入到装有磁子的瓶子中,加入50mln,n-二甲基甲酰胺,搅拌溶解均匀后将反应混合物脱除氧气,随后将反应体系放置在13w波长为400-750nm的白色led灯照射下反应24个小时,通过核磁共振氢谱测得2-(三氟甲基)乙烯基硼酸频哪醇酯转化率为99%,乙烯基正丁醚转化率为95%,聚合物样品用体积比为1:1的甲醇和水混合溶液沉淀三次,真空干燥至恒重,得到白色固体,通过凝胶渗透色谱测得的聚合物分子量mn=1.8×104g/mol和分子量分布mw/mn=1.45。
实施例7:采用反应溶液d,通过光照合成较低分子量的2-(三氟甲基)乙烯基硼酸频哪醇酯与乙烯基正丁醚共聚物
按2-(三氟甲基)乙烯基硼酸频哪醇酯:乙烯基正丁醚:2,4,6-三甲基苯甲酰基-二苯基氧化膦:对甲苯二硫醚=10:10:0.05:1的摩尔比将上述原料50mmol、50mmol、0.25mmol、5mmol分别加入到装有磁子的瓶子中,加入50mln,n-二甲基甲酰胺,搅拌溶解均匀后将反应混合物脱除氧气,随后将反应体系放置在13w波长为400-750nm的白色led灯照射下反应24个小时,通过核磁共振氢谱测得2-(三氟甲基)乙烯基硼酸频哪醇酯转化率为56%,乙烯基正丁醚转化率为43%,聚合物样品用体积比为1:1的甲醇和水混合溶液沉淀三次,真空干燥至恒重,得到白色固体,通过凝胶渗透色谱测得的聚合物分子量mn=4.1×103g/mol和分子量分布mw/mn=1.57。
实施例8:采用反应溶液c,通过光照合成富含偕三氟甲基/硼酸频哪醇结构单元的2-(三氟甲基)乙烯基硼酸频哪醇酯与醋酸乙烯酯共聚物
按2-(三氟甲基)乙烯基硼酸频哪醇酯:醋酸乙烯酯:2,4,6-三甲基苯甲酰基-二苯基氧化膦=900:100:1的摩尔比将上述原料90mmol、10mmol、0.1mmol分别加入到装有磁子的瓶子中,加入50mln,n-二甲基甲酰胺,搅拌溶解均匀后将反应混合物脱除氧气,随后将反应体系放置在13w波长为400-750nm白色led灯照射下反应24个小时,通过核磁共振氢谱测得2-(三氟甲基)乙烯基硼酸频哪醇酯转化率为32%,醋酸乙烯酯转化率为99%,聚合物样品用体积比为1:1的甲醇和水混合溶液沉淀三次,真空干燥至恒重,得到白色固体,通过聚合物的1hnmr谱图分析得到,相比于醋酸乙烯酯结构单元,偕三氟甲基/硼酸频哪醇酯结构单元在聚合物中的摩尔分数为0.87;通过凝胶渗透色谱测得的聚合物分子量mn=5.2×103g/mol和分子量分布mw/mn=1.71。
实施例9:采用反应溶液c,通过光照合成2-(三氟甲基)乙烯基硼酸邻苯二酚酯/醋酸乙烯酯/n-乙烯基吡咯烷酮的三元聚合物
按2-(三氟甲基)乙烯基硼酸频哪醇酯:醋酸乙烯酯:n-乙烯基吡咯烷酮:2,4,6-三甲基苯甲酰基-二苯基氧化膦=100:100:100:1的摩尔比将上述原料50mmol、50mmol、50mmol、0.5mmol分别加入到装有磁子的瓶子中,加入50mln,n-二甲基甲酰胺,搅拌溶解均匀后将反应混合物脱除氧气,随后将反应体系放置在13w波长为400-750nm白色led灯照射下反应24个小时,通过核磁共振氢谱测得2-(三氟甲基)乙烯基硼酸频哪醇酯转化率为99%,醋酸乙烯酯转化率为86%,n-乙烯基吡咯烷酮的转化率为91%,聚合物样品用体积比为1:1的甲醇和水混合溶液沉淀三次,真空干燥至恒重,得到白色固体,通过凝胶渗透色谱测得的聚合物分子量mn=9.2×103g/mol和分子量分布mw/mn=1.98。
第三部分:合成新型羟基官能化氟聚合物
实施例10:将5g实施例2中得到的新型硼酸酯官能化氟聚合物,加入到带有搅拌子的圆底瓶中,向烧瓶中加入100ml四氢呋喃,使硼酸酯官能化氟聚合物完全溶解,缓慢向烧瓶中加入50ml浓度为2mol/l的氢氧化钠溶液,充分搅拌均匀后,再缓慢向烧瓶中滴加50ml质量分数为30%的双氧水溶液。常温搅拌反应6个小时后,加入1mol/l盐酸溶液进行酸化,加入乙醚进行萃取,将有机相合并,减压蒸馏除去有机溶剂,真空干燥至恒重,得到新型羟基官能化氟聚合物,其1hnmr谱图、19fnmr谱图分别如图5(a)、(b)所示。通过1hnmr谱图分析可以看出,在氧化水解后,硼酸频哪醇酯上四个甲基对应的化学位移1.25ppm处的信号峰消失,表明硼酸频哪醇酯官能团的氧化转化。从19fnmr谱图可以看出,三氟甲基的信号峰从原来的化学位移62.9ppm移动到80.1ppm,且谱图上并未出现其他新的信号峰,表明硼酸频哪醇酯基团完全被氧化成羟基,得到偕三氟甲基/羟基氟聚合物。通过凝胶渗透色谱测得的聚合物分子量mn=6.5×103g/mol和分子量分布mw/mn=1.52。
第四部分:合成新型同时含有硼酸酯基团与羟基的氟聚合物
实施例11:将5g实施例2中得到的新型硼酸酯官能化氟聚合物,加入到带有搅拌子的圆底瓶中,向烧瓶中加入100ml四氢呋喃,使硼酸酯官能化氟聚合物完全溶解,缓慢向烧瓶中加入25ml浓度为2mol/l的氢氧化钠溶液,充分搅拌均匀后,再缓慢向烧瓶中滴加50ml质量分数为30%的双氧水溶液。常温搅拌反应10分钟后,加入1mol/l盐酸溶液进行酸化,加入乙醚进行萃取,将有机相合并,减压蒸馏除去有机溶剂,真空干燥至恒重,得到新型同时含有硼酸酯基团与羟基的氟聚合物,19fnmr谱图如图6所示。通过19fnmr测得该聚合物中硼酸酯基团:羟基=1:3。通过凝胶渗透色谱测得的聚合物分子量mn=7.2×103g/mol和分子量分布mw/mn=1.51。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!