一种新型组织单细胞空间转录组技术
本发明涉及生物
技术领域:
,具体地,本发明涉及一种用于检测非解离状态细胞的单细胞空间转录组技术。
背景技术:
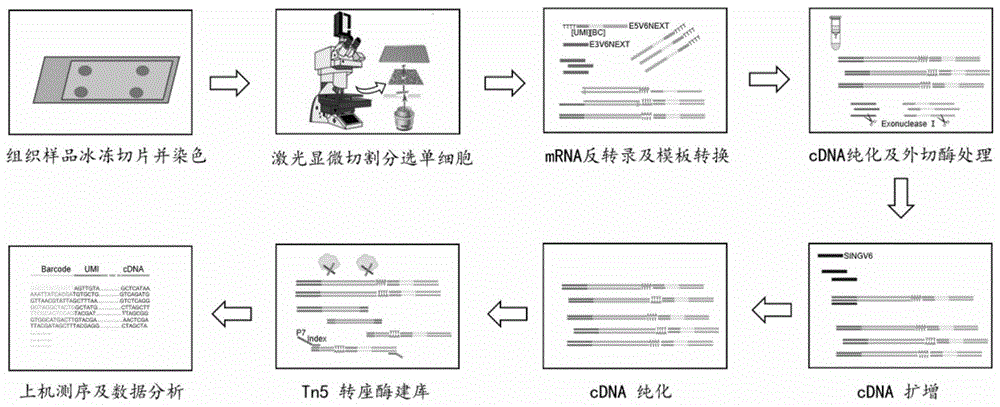
:对正常和疾病组织中的细胞进行基因表达的定量测量和研究,是理解正常组织功能和疾病组织异常的关键信息。近年来单细胞转录组技术的快速发展和应用,已经突破传统转录组技术只能获得大量细胞平均基因表达信息的局限,提供了解析单个细胞基因表达谱的能力,使得在单细胞分辨率进一步解析各种生物学问题成为可能。多种单细胞转录组文库构建方法层出不穷,大大提高了单细胞转录组测序技术的通量,灵敏度和准确性,使其在发育生物学、微生物学、神经科学等领域发挥着越来越重要的作用,成为生命科学研究的热点。在多种单细胞转录组文库构建方法中,单细胞rna条码测序方法(scrb-seq,us14898030a1)由于检测灵敏度高,成本低廉,方法灵活性强等优点,吸引了研究者的兴趣。该方法基于中等通量的96/384孔板,采用细胞标签(barcode)和转录本标签umi(uniquemolecularidentifier)的富集3'端建库策略。细胞barcode的使用提高了单细胞放大的均一性和重复性,umi的使用可以消除pcr扩增偏差,实现对转录本的定量分析。并且,该方法使用设备简单,适合在实验室推广。因此,这一方法在单细胞转录组分析中具有巨大的应用潜力。目前用单细胞转录组技术(包括scrb-seq技术)对组织中细胞进行基因表达分析,需要将组织酶解成单细胞悬液进行,如目前常用的流式细胞分选、口吸管法和微流控系统分离等技术,这导致细胞在组织中原本的空间位置信息丢失,从而无法将基因表达的结果与细胞的具体位置联系起来,而细胞的空间位置对于理解细胞的分化命运、细胞间的相互作用等具有重要意义。另一方面,较长时间的酶解消化过程,以及口吸管产生的环境压力等会对细胞产生强烈刺激,可能引起细胞产生应激反应,使转录组信息发生改变,甚至导致特异性细胞类型的rna选择性降解,进而影响转录组信息的准确性。因此,基于组织原位的空间信息,对组织中的细胞进行基因表达测量,是理解细胞在组织真实微环境中行为和功能的关键,对于理解组织发育、疾病发生等具有重要意义。近年来,空间转录组学研究的重要性得到越来越多研究者的重视。现有的空间转录组技术主要分为两类:一类是基于rna荧光原位杂交杂交和成像的方法,如smfish,merfish,seqfish等;另一类是基于测序的方法,包括tiva,iss,fisseq,以及基于激光显微切割(lasercapturemicrodissection,lcm)的细胞转录组分析方法等。smfish,seqfish等方法在分析的细胞数量和检测靶点的数量上都很有限,并且需要大量的fish探针,连续杂交过程也很耗时;而tiva等基于测序的方法虽然可分析的细胞数量有提高,但转录本检测效率普遍较低。与上述方法相比,如果将激光显微切割技术和高通量的单细胞转录组测序技术相结合,既可以获得细胞的空间信息,又能同时获得大量基因表达的定量信息。激光显微切割技术采用对核酸无损伤的激光精准捕获组织切片中的目标细胞,组织样品前处理时以液氮速冻代替酶消化解离,避免了传统单细胞分离时因较长时间酶解、机械压力等对细胞转录组造成的影响,因此可以最大程度地保留细胞原始的空间位置信息和真实的转录组信息。将获得的转录本信息最终对应回组织内原始位置,可以最大程度的还原组织内部各空间位置的基因表达状态。虽然将激光显微切割获得的样品用于大量细胞转录组测序已有很多报道,但是这些研究往往需要收集几十至数千个细胞。因此,本领域仍迫切需要开发一种既可以获得细胞的空间信息,又能够实现对单细胞转录组高通量测序,且灵敏度高和一致性好的方法。技术实现要素:本发明的目的在于提供一种检测非解离状态细胞的单细胞空间转录组技术。本发明的第一方面,提供了一种单细胞空间转录组分析方法,所述方法包括以下步骤:(1)提供一待测样品,所述待测样品为组织样品,将所述待测样品在z轴方向进行冰冻切片处理,从而获得n个组织切片,其中所述组织切片设有相应的z轴坐标值,并且所述组织切片厚度d1与所述组织样品中对应细胞的平均细胞直径dc满足1/2dc≤d1≤2dc;(2)对于所述组织切片,分别进行激光显微切割,从而从所述组织切片上获得m个单个目标细胞,其中,每个目标细胞设有相应的三维坐标值(xi,yi,zi),式中,i为1至m,并且m≥500(较佳地m为1000-106);(3)对于所述的每个目标细胞,进行裂解,释放mrna,并进行cdna文库构建,从而得到对应于每个目标细胞的cdna文库;(4)对所述目标细胞cdna文库测序分析,从而获得每个目标细胞的单细胞空间转录组测序数据;和(5)基于每个目标细胞的三维坐标值,将每个目标细胞的单细胞空间转录组测序数据映射于组织样品的对应空间上,从而获得所述组织样品的单细胞空间转录组测序数据集。在另一优选例中,所述组织样品来源于离体组织。在另一优选例中,所述组织样品来源于人类或非人类动物。在另一优选例中,所述组织样品是利用液氮进行组织速冻的样品。在另一优选例中,所述组织切片厚度d1与所述组织样品中对应细胞的平均细胞直径dc满足1/2dc≤d1≤dc。在另一优选例中,所述组织切片的厚度d1为7-25μm。在另一优选例中,在步骤(2)中,还包括以下步骤:(a)将所述组织切片贴于载玻片上;(b)对所述组织切片固定后进行快速染色,从而得到染色后的组织切片;(c)利用激光显微切割系统,根据染色后的样品中细胞的形态圈出单个目标细胞并进行激光切割,从而收集单个目标细胞至收集管,所述收集管内含预先加入的细胞收集液,所述细胞收集液包含细胞裂解液和带有细胞标签(barcode)的引物;和(d)将收集到的目标细胞密封在收集管中在-80℃条件下冷冻保存。在另一优选例中,在步骤(a)中,所述载玻片为经冰预冷过的载玻片。在另一优选例中,在步骤(a)中,所述载玻片为经冰预冷过的pen膜玻片。在另一优选例中,所述“经冰预冷”是指提前将载玻片装入50ml离心管中插在冰上(0℃)预冷。在另一优选例中,在步骤(b)中,所述组织切片用95%乙醇固定,固定时间为0.5-2分钟。在另一优选例中,在步骤(b)中,所述快速染色包括:甲基紫染色或甲基紫—伊红染色在另一优选例中,在步骤(b)中,所述染色的时间为1-3分钟,染色的时间需根据组织类型和组织切片厚度进行调整。在另一优选例中,在步骤(b)中,在染色后,还进一步包括对染色后的组织切片进行脱水干燥的步骤。在另一优选例中,所述脱水干燥为用浓度梯度的乙醇进行乙醇梯度脱水干燥,依次为95%乙醇两次,100%乙醇三次。在另一优选例中,在步骤(c)中,所述收集管为无核酸酶的收集管。在另一优选例中,在步骤(c)中,所述收集管为单个无核酸酶的pcr管,或具有n个孔的细胞收集板中的一个孔,其中n为≥2的正整数,较佳地,n为96或384。在另一优选例中,所述收集管为96孔细胞收集板中的一个孔。在另一优选例中,在步骤(c)中,所述细胞裂解液中含有tritonx-100,其在细胞收集液中的终浓度为0.1-0.2%,较佳地为0.2%。在另一优选例中,在步骤(c)中,所述细胞裂解液中进一步含有rna酶抑制剂,其在细胞收集液中的终浓度为1-2u/μl,较佳地为2u/μl。在另一优选例中,在步骤(c)中,所述带有细胞标签(barcode)的引物为含有6bp细胞标签(barcode)序列和10bp随机分子标签(umi)序列的引物,所述引物具有如seqidno:1所示的序列。在另一优选例中,所述6bp细胞标签(barcode)序列在每个收集管(孔)中的序列不同,彼此之间至少有两个碱基的差异。在另一优选例中,在步骤(c)中,所述带有细胞标签(barcode)的引物加入细胞收集液前的浓度为2μm。在另一优选例中,在步骤(3)中,所述裂解包括以下步骤:将冷冻保存的含有目标细胞的收集管置于冰上(0℃)融化10-20分钟。在另一优选例中,在步骤(3)中,所述cdna文库构建包括以下步骤:(i)以目标细胞释放的mrna为模板进行逆转录反应,从而得到含cdna的逆转录产物;(ii)对步骤(i)中所述逆转录产物进行纯化后并用外切酶处理,从而得到外切酶处理的cdna;(iii)对所述外切酶处理的cdna进行pcr扩增,从而得到经扩增的cdna;和(iv)对所述经扩增的cdna进行纯化后使用dna建库试剂盒进行建库,从而得到所述目标细胞的cdna文库。在另一优选例中,在步骤(iv)中,所述cdna建库试剂盒选自下组:诺唯赞的trueprepdnalibraryprepkit或illumina的nexteraxtdnalibrarypreparationkit。在另一优选例中,在步骤(4)中,所述测序的方法选自下组:illuminahiseq或novaseq系统测序。在另一优选例中,所述的方法用于非诊断目的。本发明的第二方面,提供了一种单细胞空间转录组分析系统,所述系统包括:(m1)冷冻切片模块,所述模块被配置为:将待测组织样品在z轴方向进行冰冻切片处理,从而获得n个组织切片,其中所述组织切片设有相应的z轴坐标值,并且所述组织切片厚度d1与所述组织样品中对应细胞的平均细胞直径dc满足dc<d1≤2dc;(m2)激光显微切割模块,所述模块被配置为:对所述组织切片分别进行激光显微切割,从而从所述组织切片上获得m个单个目标细胞,其中,每个目标细胞设有相应的三维坐标值(xi,yi,zi),式中,i为1至m,并且m≥500(较佳地m为1000-106);(m3)转录组学测序模块,所述模块被配置为:对所述每个目标细胞进行cdna文库构建,从而得到对应于每个目标细胞的cdna文库;然后对所述目标细胞cdna文库测序分析,从而获得每个目标细胞的单细胞空间转录组测序数据;和(m4)分析模块,所述模块被配置为:基于每个目标细胞的三维坐标值,将每个目标细胞的单细胞空间转录组测序数据映射于组织样品的对应空间上,从而获得所述组织样品的单细胞空间转录组测序数据集。在另一优选例中,所述的分析系统还包括:(m5)控制模块,用于控制模块(m1)冷冻切片模块、(m2)激光显微切割模块、(m3)转录组学测序模块、和(m4)分析模块的运作。在另一优选例中,所述的控制模块控制所述单细胞空间转录组分析系统中的以下模块依次运作:(m1)冷冻切片模块、(m2)激光显微切割模块、(m3)转录组学测序模块、和(m4)分析模块,即按照按(m1)→(m2)→(m3)→(m4)的顺序依次运行。在另一优选例中,所述(m2)激光显微切割模块进一步包含以下子模块:(m2-1)切割前处理模块和(m2-2)切割模块;其中,所述(m2-1)切割前处理模块被配置为:将所述组织切片贴于经冰预冷过的载玻片上,然后对所述组织切片固定后进行快速染色,从而得到染色后的组织切片;所述(m2-2)切割模块被配置为:利用激光显微切割系统,根据染色后的样品中细胞的形态圈出单个目标细胞并进行激光切割,从而收集单个目标细胞至收集管。在另一优选例中,所述(m3)转录组学测序模块进一步包含以下子模块:(m3-1)裂解模块、(m3-2)cdna建库模块和(m3-3)测序模块;其中,所述(m3-1)裂解模块被配置为:将在-80℃条件下冷冻的含有目标细胞的收集管置于冰上10-20分钟,从而得到含有目标细胞mrna的裂解产物;所述(m3-2)cdna建库模块被配置为:利用所述含有目标细胞mrna的裂解产物,对收集的目标细胞进行cdna文库构建,从而得到目标细胞cdna文库;所述(m3-3)测序模块被配置为:对所述目标细胞cdna文库进行测序。在另一优选例中,所述的控制模块控制所述(m2)激光显微切割模块的子模块按照(m2-1)→(m2-2)的顺序依次运行。在另一优选例中,所述的控制模块控制所述(m3)转录组学测序模块的子模块按照(m3-1)→(m3-2)→(m3-3)的顺序依次运行。本发明的第三方面,提供了一种单细胞转录组分析方法,所述方法包括以下步骤:(a)提供一待测样品,所述样品为含有细胞的液态样品,将所述样品涂布于载玻片上,从而得到贴附于载玻片上的细胞样品;(b)对所述贴附于载玻片上的细胞样品固定后进行快速染色,从而得到染色后的样品;(c)利用激光显微切割系统,根据染色后的样品中细胞的形态圈出n个单个目标细胞并进行激光切割,从而分别收集每个目标细胞至n个收集管,所述收集管内含预先加入的细胞收集液,所述细胞收集液包含细胞裂解液和带有细胞标签的引物;(d)对步骤(c)收集的目标细胞进行裂解,从而得到含有目标细胞mrna的裂解产物;(e)利用所述含有目标细胞mrna的裂解液,对所述目标细胞进行cdna文库构建,从而得到目标细胞cdna文库;和(f)对所述目标细胞cdna文库测序分析,从而对所述样品进行单细胞转录组分析。应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。附图说明图1显示了lcm-scrb-seq方法的实验流程图。图2显示了应用lcm-scrb-seq方法得到的30天小鼠卵母细胞全长cdna的agilent2100分析峰图。图3显示了应用lcm-scrb-seq方法对30天小鼠的卵母细胞进行单细胞测序所得基因数分布。图4显示了利用lcm-scrb-seq方法对同步化到中期的hela细胞进行单细胞测序所得单个细胞间基因表达的相关性分布:(a)所有细胞总体的基因表达相关性分布图;(b)六对代表性单细胞的基因表达分布散点图。图5显示了激光显微切割分选卵母细胞过程图:(a)细胞切割前;(b)细胞切割完成弹射收集。图6显示了scrb-seq方法的建库流程图:首先在单个管中实现细胞mrna的反转与加标签,再将所有管中溶液合并到一起进行cdna过柱纯化,然后使用外切酶i消化多余的引物,再通过pcr试剂盒扩增cdna,纯化后使用转座酶法建库,应用illuminahiseq系统测序后进行数据分析。图7显示了通过本发明的lcm-scrb-seq方法获得的30天小鼠的卵母细胞cdnaagilent2100分析峰图(a)及基因数分布(b)。图8显示了通过本发明的lcm-scrb-seq方法获得的同步化hela细胞cdnaagilent2100分析峰图(a)及基因数分布(b)。图9显示了利用本发明的lcm-scrb-seq方法进行卵巢组织三维空间重构的流程图。具体实施方式本发明人经过广泛而深入的研究,首次开发了一种可以检测非解离状态的单细胞空间转录组技术。具体地,本发明的方法和装置可在保有组织中细胞空间位置信息的基础上,高通量地收集单细胞,从而对组织中具有空间分布的单细胞进行空间转录组分析,并对基于空间转录组分析数据进行三维重构,从而为组织中细胞状态和功能研究提供更高效和准确的工具。本发明方法具有高灵敏性、高重复性和可定量化等优点,可广泛应用于正常和疾病组织的单细胞基因表达定量研究。在此基础上,完成了本发明。术语激光显微切割如本文所用,术语“激光纤维切割”即激光捕获显微切割(lasercapturemicrodissection(lcm)),是在不破坏组织结构,保存要捕获的细胞和其周围组织形态完整的前提下,直接从冰冻或石蜡包埋组织切片中获取目标细胞。在本发明中,切割得到的目标细胞可以被收集到带粘性盖子的收集管中。在另一优选例中,所述收集管为单个无核酸酶的pcr管,或96孔的细胞收集板。在本发明中,在以下条件下进行激光显微切割:(1)环境湿度在45%以下;(2)在可以正常切割和弹射的基础上,尽可能采用最低的切割能量和弹射能量。具体地,可以设置以下参数进行激光显微切割:10倍镜下,cutenergy:47-51;cutfocus:82-84;laserenergy:80-83;laserfocus:82-84。实际操作中参数需依据组织特性进行调整。单细胞rna条码测序(scrb-seq)scrb-seq(singlecellrnabarcodingsequencing)作为高通量转录组方法,基于较高通量的96/384孔板,采用富集3'端的策略,使用barcode序列对细胞进行标记,并整合特异性的分子条码umi,以分辨扩增分子的来源,更准确的定量转录本。本发明的方法中所用的scrb-seq试剂和材料可参见(us14898030a1),具体地,本发明所用的scrb-seq试剂如下:(1)细胞标签引物e3v6next(seqidno:1):5'-/5biosg/acactctttccctacacgacgatct[bc6]nnnnnnnnnnttttttttttttttttttttttttttttttvn-3';其中,5biosg为5'biotin;v=a或g或c,n=a或g或c或t;[bc6]为6bpbarcode序列,由atcg四种脱氧核苷酸组成,其在每个收集管(孔)中的序列不同,barcode彼此之间至少有两个碱基的差异,因此单个碱基测序错误不会导致barcode的错误识别;(n)10为10bp随机分子标签(uniquemolecularidentifier(umi)),每个umi被整合到一个由细胞mrna生成的独特cdna序列中,以分辨扩增分子的来源。(2)逆转录引物e5v6next(seqidno:2):5'-icigicacactctttccctacacgacgcrgrgrg-3';其中,ic为iso-dc(锁核酸修饰的c碱基);ig为iso-dg;rg为rnag(即核糖核苷酸g)。(3)cdna扩增引物singv6primer(seqidno:3):5'-/5biosg/acactctttccctacacgacgc-3';其中,5biosg为5'biotin。(4)i5接头p5nextpt5(seqidno:4):5'-aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccg*a*t*c*t*-3';其中,*表示硫代磷酸酯键(phosphorothioatebond)。本发明的方法本发明提供了一种可以检测非解离状态的单细胞空间转录组技术,所述方法包括以下步骤:(1)提供一待测样品,所述待测样品为组织样品,将所述待测样品在z轴方向进行冰冻切片处理,从而获得n个组织切片,其中所述组织切片设有相应的z轴坐标值,并且所述组织切片厚度d1与所述组织样品中对应细胞的平均细胞直径dc满足1/2dc<d1≤2dc,较佳地,所述组织切片厚度d1与所述组织样品中对应细胞的平均细胞直径dc满足1/2dc≤d1≤dc。(2)对于所述组织切片,分别进行激光显微切割,从而从所述组织切片上获得m个单个目标细胞,其中,每个目标细胞设有相应的三维坐标值(xi,yi,zi),式中,i为1至m,并且m≥500(较佳地m为1000-106);(3)对于所述的每个目标细胞,进行裂解,释放mrna,并进行cdna文库构建,从而得到对应于每个目标细胞的cdna文库;(4)对所述目标细胞cdna文库测序分析,从而获得每个目标细胞的单细胞空间转录组测序数据;和(5)基于每个目标细胞的三维坐标值,将每个目标细胞的单细胞空间转录组测序数据映射于组织样品的对应空间上,从而获得所述组织样品的单细胞空间转录组测序数据集。如本文所用,术语“非解离状态的单细胞”主要是指非悬浮和非游离状态的单细胞,例如,在组织中的单细胞,其未经解离(例如酶解消化),具有相对确定的空间位置。在本发明的优选例中,“非解离状态的单细胞”可以是位于离体组织中的单个细胞。本发明将scrb-seq(参见us14898030a1)技术进一步发展,应用于组织中单细胞的转录组研究,建立了一种新的三维空间基因组的方法(lcm-scrb-seq)。本发明的方法主要优化之处如下:(1)和传统的scrb-seq方法相比,lcm-scrb-seq方法组织离体后无需将细胞酶解,立即放入液氮速冻,避免细胞酶解可能引起的转录组变化,保持了组织细胞的转录组真实性。(2)组织在单细胞分选之前,对组织进行冰冻切片后,用预冷过的95%乙醇固定,可以较好的保持组织形态和保护rna质量。之后,采用快速甲基紫染色的方法对组织中细胞进行染色,尽可能减少组织切片在染色液及外界环境中暴露的时间,防止rna降解。(3)在激光显微切割中采用96孔板收集装置,实现较高通量的单细胞选择。(4)通过在细胞收集缓冲液组分中增加终浓度为0.2%的tritonx-100,一方面提高组织中激光显微切割分选单细胞的收集率,另一方面提高组织细胞的裂解效率。本方法在细胞分选开始前,预先向收集管中加入配制好的细胞收集缓冲液,缓冲液中的tritonx-100可以有效降低水的表面张力,使得细胞收集液更容易铺满收集盖,有效提高激光显微切割后的细胞的收集率。同时,在细胞裂解过程中,tritonx-100也提高了细胞的裂解效率。(5)为了有效提高激光显微切割收集后的细胞rna质量,本方法在细胞收集缓冲液组分中增加终浓度为2u/μl的rna酶抑制。rna酶抑制剂的添加可以使细胞在进入液体环境后,内源性rna酶活性被抑制,rna得到有效保护。本发明的系统本发明还提供了一种可人工控制或通过仪器自动化进行的单细胞空间转录组分析系统(或装置),该系统可以对离体组织样品进行冰冻切片处理、组织切片固定和染色、激光显微切割、细胞裂解、cdna建库以及测序分析,从而获得组织样品中的单细胞转录组数据。该系统可实现中高通量的组织中单细胞转录组数据采集,适用于组织中单细胞空间转录组信息采集和建库。具体地,所述所述系统(或装置)包括:(m1)冷冻切片模块;(m2)激光显微切割模块;(m3)转录组学测序模块;(m4)分析模块;和(m5)控制模块。所述的控制模块控制所述单细胞空间转录组分析系统中的以下模块依次运作:(m1)冷冻切片模块、(m2)激光显微切割模块、(m3)转录组学测序模块、和(m4)分析模块,即按照按(m1)→(m2)→(m3)→(m4)的顺序依次运行。本发明的系统还可进一步包括计算机、数据存储器、降温、加温和恒温孵育装置以及其他连接部件,以组成完整的系统。本发明的主要优点包括:(1)实现了单细胞分辨率的空间转录组分析:本发明的方法采用组织原位的激光显微切割,无需消化成细胞悬液可直接在组织切片上进行细胞分选,不仅可以对组织形貌进行直接观察,便于识别记录细胞原有的空间位置,还减少了酶解消化等步骤,尽可能降低了外界酶解及温度的长时间刺激对细胞转录组的影响。(2)获得了高通量的组织单细胞转录组数据:本发明的方法采用96孔的收集板,在建库中对细胞进行barcode标记后,将单细胞的转录本文库混合进行后续操作,大大提高了实验的通量,以及单细胞转录组数据的重复性。(3)从组织原位捕获到的单细胞中获得高质量的cdna:本发明的方法获得的长度超过1000bp的全长cdna占比超过50%,甚至已超过单细胞转录组金标准smart-seq2所做到的50%左右。(4)本发明的方法灵敏度较高且一致性较好。(5)本发明的方法也适用于细胞悬液中少量细胞的分选,成为传统分析方法必要的补充。下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如sambrook等人,分子克隆:实验室手册(newyork:coldspringharborlaboratorypress,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。本发明中所涉及的实验材料和试剂如无特殊说明均可从市售渠道获得。实施例1本发明的lcm-scrb-seq方法1.细胞收集液的制备按照如下组分配制缓冲液(含稀释500倍的phusionhfbuffer,2u/μlrna酶抑制剂,0.2%tritonx-100)。表1试剂体积phusionhfbuffer(newenglandbiolabs,usa)2μlrnaseinhibitor35.8μl2%tritonx-10071.6μlnuclease-freewater606.6μl总体积716μl在单个无核酸酶的pcr管中加入10μl细胞收集液(8μl缓冲液和2μl带有细胞标签的引物(e3v6next,us14898030a1),引物使用浓度为2μm),或在lcm96孔收集板的板盖内每孔加入7μl细胞收集液(5μl缓冲液和2μl带有细胞标签的引物,浓度为1μm),盖上96孔板,板盖在下放入4℃冰箱备用。2.冰冻切片染色取出冷冻状态的组织,冰冻切片10-25μm(据所要分选的细胞体积选择),将组织切片贴在经冰中预冷过的带膜pen玻片上,放入95%乙醇固定2min,甲基紫染液染色1-2min(染色时间需要根据组织特性及切片厚度进行合理调整),乙醇梯度脱水干燥后将玻片自然风干几秒,用手稍微平衡温度,避免组织暴露在外界环境中再次吸水。3.细胞收集提前打开除湿机将激光显微切割仪房间的湿度降至45%以下,将仪器提前开机并校准,调好参数(10倍镜下,cutenergy:47-51;cutfocus:82-84;laserenergy:80-83;laserfocus:82-84,实际参数需依据组织特性进行调整),在可以正常切割和弹射的基础上,尽可能采用最低的切割能量和弹射能量。调整好显微镜焦距,根据染色后的组织形态圈出单个目标细胞并进行激光切割。对于单个pcr管收集细胞,将提前配制好的10μl裂解液转入用于收集的单个pcr管的管盖中,固定在lcm8×200μl收集板架上,调整好管盖的位置,使其正对显微镜视野中心,进行激光弹射收集细胞,每个管盖中分别收集一个目标细胞。分选结束后取下收集有细胞的8个pcr管,立即转入-80℃冰箱。对于lcm96孔板收集细胞,将每孔提前加有7μl细胞收集液的96孔板的板盖置于lcm收集96孔板收集架上,调整好孔的位置,使每个孔在收集细胞时都正对显微镜视野中心,每孔分别收集一个目标细胞。收集结束后用自封袋将96孔板密封,立即转入-80℃冰箱。(此处的目的既是暂时储存不能立即检测的样品,也是为后续通过冻融裂解细胞做准备)4.细胞裂解将收集有细胞的lcm96孔板(或pcr管)从-80℃冰箱中取出,冰上(0℃)放置约10min融化后,4℃,4000rpm离心5min,将样品收集至管底(沉淀于管底的是mrna以及细胞碎片)。在超净台中打开96孔板(或pcr管)的盖子70℃敞口加热蒸发至溶液剩余2μl,置于冰盒上。5.mrna反转录为cdna使用maximahminusreversetranscriptase(thermoscientific,#ep0753),配制反应体系如下(96孔板量):表2试剂体积5xrtbuffer100μldntps50μlnuclease-freewater125μle5v6next(us14898030a1)adapter4μlrnaseoutrecombinantrnaseinhibitors6μlmaximahminusreversetranscriptase10μl总体积295μl每孔加入3μl上述混合反应液,用粘性封口膜替换掉lcm96孔收集板的盖子,密封好,置于pcr仪42℃孵育90min。6.cdna纯化及外切酶作用将lcm96孔板(或pcr管)每孔中的溶液全部汇集,使用zymoresearchdna纯化试剂盒(#d4013)纯化cdna,具体操作为:(1)将96孔板中的溶液汇入到1.5ml离心管中,加入7倍体积的dnabindingbuffer,快速混匀;(2)将混匀后的溶液转入套有收集管的zymo-spin吸附柱中;(3)10000g离心30s,丢弃收集管中的溶液;(4)向吸附柱中加入200μldnawashbuffer,1000g离心30s;(5)重复步骤(4)一次;(6)加入18μlnuclease-freewater于吸附柱中,室温孵育1min,后将吸附柱转入新的离心管中,1000g离心30s,洗脱cdna;(7)加入2μl10×reactionbuffer和1μlofexonucleasei,37℃孵育30min,80℃孵育20min。7.cdna放大使用terrapcrdirectpolymerasemix(clontech,#639270)试剂盒进行cdna放大,配置如下反应体系:表3试剂体积前述步骤的cdna20μl2xterrapcrdirectbuffer25μlsingv6primer(us14898030a1)1μlterrapcrdirectpolymerasemix1μlnuclease-freewater3μl总体积50μl充分吹打混匀后放入pcr仪,按照如下参数设置pcr扩增程序:表4反应结束后,使用0.8倍体积的bechmanampurexp磁珠纯化扩增后的cdna,使用qubit3.0荧光定量仪测定浓度后用安捷伦2100核酸分析仪检测cdna质量。8.建库纯化后的cdna用trueprepdnalibraryprepkitv2forillumina(诺唯赞,#td502)试剂盒构建测序文库,具体操作如下:(1)在灭菌pcr管中配制如下反应体系:表5试剂体积5×ttbl4μl5ngcdnaxμlttemixv55μlddh2o至20μl多次吹打充分混匀,将反应管置于pcr仪中,运行以下程序:表6105℃热盖55℃10min10℃hold反应结束后立即向产物中加入5μl5×ts,使用移液器轻轻吹打充分混匀,室温放置5min。(2)pcr富集将pcr管放冰上,使用p5nextpt5(us14898030a1)作为i5接头,其中n7xx*为建库时使用的index,配置如下反应体系:表7试剂体积p5nextpt55μln7xx*5μltae1μl5×tab10μlddh2o4μl总体积25μl将以上配制的25μl反应液加入上一步反应结束的pcr管中,得到终体积为50μl的反应体系,多次吹打充分混匀后置于pcr仪中,运行以下程序:表8(3)扩增产物长度分选使用bechmanampurexp磁珠对扩增产物进行片段长度分选,qubit检测浓度后使用安捷伦2100检测文库质量,文库片段分布应在300-800bp。9.高通量测序用illuminanovaseq系统测序。实施例2本发明的lcm-scrb-seq方法的特性分析1.从组织原位捕获到的单细胞中cdna质量检测如实施例1所述,使用本发明的lcm-scrb-seq方法进行激光显微切割分选单细胞,mrna反转,cdna扩增并纯化后,得到的cdna进行agilent2100分析质检。图2质检结果显示,本发明的lcm-scrb-seq方法获得的长度超过1000bp的全长cdna占比超过50%,甚至已超过单细胞转录组金标准smart-seq2所做到的50%左右,满足建库要求。2.灵敏度检测应用本发明的lcm-scrb-seq方法(如实施例1所述)对30天正常小鼠的卵母细胞进行建库测序。结果显示,在每个细胞1mreads的测序深度下,每个细胞测到的平均基因数为10320(图3),与之前已发表的应用smart-seq对小鼠单个卵母细胞转录组测序测得的基因数基本相当,说明本发明的lcm-scrb-seq方法的灵敏度较高。3.一致性检测将hela细胞同步化到有丝分裂中期,获得基因表达相对均一的细胞,利用本发明的lcm-scrb-seq方法(如实施例1所述)通过激光显微切割将单个贴在玻片上的细胞分选出来建库测序。得到测序结果后将每个细胞中的转录本数进行标准化,并计算单个细胞之间基因表达的相关性。结果如图4所示,发现单个细胞之间基因表达的相关性较高,平均在0.87以上(图4a)。表明细胞之间由于方法本身造成的差异较小,本发明的lcm-scrb-seq方法的一致性较好。实施例3应用本发明的lcm-scrb-seq方法对30天小鼠卵母细胞进行单细胞转录组测序1.细胞收集液的制备在lcm96孔收集板的板盖内每孔加入7μl细胞收集液(5μl缓冲液和2μl带有细胞标签的引物,浓度为1μm),缓冲液按照实施例1中表1配制,盖上96孔板,板盖在下放入4℃冰箱备用。2.冰冻切片并染色从-80℃取出30天小鼠卵巢组织,冰冻切片20μm,将组织切片贴在经冰中预冷过的带膜pen玻片上,放入95%乙醇固定2min,甲基紫染液染色1min50s,乙醇梯度脱水干燥后将玻片自然风干几秒,用手稍微平衡温度。3.细胞收集提前打开除湿机将激光显微切割仪房间的湿度降至45%以下,将仪器提前开机并校准,调好参数,由于卵母细胞较大,故在10倍镜下分选细胞(cutenergy:47-51;cutfocus:82-84;laserenergy:80-83;laserfocus:82-84)。调整好显微镜焦距,根据染色后的组织形态圈出单个卵母细胞并进行激光切割(图5)。将加有细胞收集液的lcm96孔板的板盖置于收集架上,调整好孔的位置,使每个孔在收集细胞时都正对显微镜视野中心,每孔分别收集一个卵母细胞。收集结束后用自封袋将96孔板密封,立即转入-80℃冰箱。4.细胞裂解将收集有细胞的lcm96孔板(或pcr管)从-80℃冰箱中取出,冰上放置10min融化后,4℃,4000rpm离心5min,将细胞收集至管底。在超净台中打开96孔板的盖子70℃敞口加热蒸发至溶液剩余2μl,置于冰盒上。5.rna反转为cdna使用thermoscientific的maximahminusreversetranscriptase(#ep0753)按照实施例1中表2配制反应体系(96孔板量)。每孔加入3μl,即共为5μl反应体系,42℃孵育90分钟。6.cdna纯化及放大将所有pcr管子中的溶液汇总到一个管子中,使用zymoresearch(#d4013)dna纯化试剂盒,最后使用18μl水洗脱cdna,加入2μl10×reactionbuffer和1μlexonucleasei,37℃孵育30分钟,80℃孵育20分钟。使用terrapcrdirectpolymerasemix(clontech,#639270)试剂盒进行cdna放大,按照实施例1中表3配制反应体系。按照实施例1中表4所示的程序进行pcr扩增后4℃保存。使用0.8xbechmanampurexp磁珠纯化扩增后的cdna。7.建库纯化后的cdna应用诺唯赞的trueprepdnalibraryprepkit进行建库,使用p5nextpt5(us14898030a1)作为i5接头,扩增10个循环,使用0.8xbechmanampurexp磁珠纯化后使用安捷伦2100核酸分析仪进行片段分析,文库在300-800bp,后应用illuminanovaseq系统测序。本实施例中的建库流程如图6所示。8.实验结果由图7a可见,应用本发明的lcm-scrb-seq方法所得的卵母细胞全长cdna片段分布主峰大于1000,后经测序获得的单个细胞平均基因产出为10320(图7b)(卵母细胞所含转录本含量比普通细胞高,故检测到更多基因)。实施例4应用本发明的lcm-scrb-seq方法对同步化到有丝分裂中期的hela进行单细胞转录组测序1.细胞收集液的制备在lcm96孔收集板的板盖内每孔加入7μl细胞收集液(5μl缓冲液和2μl带有细胞标签的引物,浓度为1μm),缓冲液按照实施例1中表1配制,盖上96孔板,板盖在下放入4℃冰箱备用。2.准备细胞悬液提前向生长密度达到80%的hela细胞中加入秋水仙素作用12-14小时,将细胞同步化至有丝分裂中期,消化细胞并重悬于灭菌pbs中,调整密度至细胞在镜下观察时较为分散,取100μl细胞悬液加入新的甩片加样管,用封口膜将管口封好,放入甩片机,400rpm,1min甩片。甩片结束在超净台中将玻片取下,自然风干几秒,放入50ml离心管,开始lcm分选。3.激光显微切割收集细胞提前打开除湿机将激光显微切割仪房间的湿度降至45%以下,将仪器提前开机并校准,调好参数,在10倍镜下分选细胞(cutenergy:45-58;cutfocus:83;laserenergy:80-83;laserfocus:83)。调整好显微镜焦距,在明场下根据细胞形态圈出单个细胞并进行激光切割。将加有细胞收集液的lcm96孔板板盖置于收集架上,调整好孔的位置,使每个孔在收集细胞时都正对显微镜视野中心,每孔分别收集一个目标细胞。收集结束后用自封袋将96孔板密封,立即转入-80℃冰箱。4.细胞裂解将收集有细胞的lcm96孔板(或pcr管)从-80℃冰箱中取出,冰上放置10min融化后,4℃,4000rpm离心5min,将细胞收集至管底。在超净台中打开96孔板的盖子70℃敞口加热蒸发至溶液剩余2μl,置于冰盒上。5.rna反转为cdna使用thermoscientific的maximahminusreversetranscriptase(#ep0753)按照实施例1中表2配制反应体系(96孔板量)。每孔加入3μl,即共为5μl反应体系,42℃孵育90分钟。6.cdna纯化及放大将所有pcr管子中的溶液汇总到一个管子中,使用zymoresearch(#d4013)dna纯化试剂盒,最后使用19μl水洗脱cdna,加入2μl10×reactionbuffer和1μlexonucleasei,37℃孵育30分钟,80℃孵育20分钟。使用terrapcrdirectpolymerasemix(clontech,#639270)试剂盒进行cdna放大,按照实施例1中表3配制反应体系。按照实施例1中表4所示的程序进行pcr扩增后4℃保存。使用0.8xbechmanampurexp磁珠纯化扩增后的cdna。7.建库纯化后的cdna应用诺唯赞的trueprepdnalibraryprepkit进行建库,使用p5nextpt5(us14898030a1)作为i5接头,扩增10个循环,使用0.8xbechmanampurexp磁珠纯化后使用安捷伦2100核酸分析仪进行片段分析,文库在300-800bp,后应用illuminanovaseq系统测序。本实施例中的建库流程如图6所示。8.实验结果图8a为同步化到中期的hela细胞全长cdna的2100分析峰图,图中可见主峰大于1000,后经测序获得的单个细胞平均基因产出为4745(图8b)。实施例5利用本发明的lcm-scrb-seq方法进行卵巢组织三维空间重构使用本发明的方法,对小鼠卵巢组织的转录组进行三维空间重构(图9),具体步骤如下:1.卵巢组织收取将30天的c57雌鼠脱颈处死后摘取卵巢,放入冰上预冷的hbss缓冲液中,在体视显微镜下去除卵巢周围的脂肪组织,将小鼠卵巢放入冻存管中,液氮速冻30分钟以上,转入-80℃冰箱。2.冰冻切片从-80℃冰箱中取出小鼠卵巢组织转入冰冻切片机,连续切片20μm,将组织切片贴在经冰中预冷过的带膜pen玻片上,放入95%乙醇固定2min,快速风干使乙醇挥发完全后转入50ml离心管中,存于-80℃冰箱备用。重复连续切片及固定操作,直至整个卵巢组织全部切完。3.细胞收集液的制备在lcm96孔收集板的板盖内每孔加入7μl细胞收集液(5μl缓冲液和2μl带有细胞标签的引物,浓度为1μm),缓冲液按照实施例1中表1配制,盖上96孔板,板盖在下放入4℃冰箱备用。4.染色从-80℃冰箱中取出切片,冰上平衡温度后放入甲基紫-伊红染液染色1min30s,乙醇梯度脱水干燥后将玻片自然风干几秒,用手稍微平衡玻片温度,防止玻片因温度过低导致水分凝结。5.细胞收集提前打开除湿机,将激光显微切割仪房间的湿度降至45%以下,将仪器提前开机并校准,调好参数,由于卵母细胞较大,故在10倍镜下进行分选(cutenergy:47-51;cutfocus:82-84;laserenergy:80-83;laserfocus:82-84)。调整好显微镜焦距,根据染色后的组织形态圈出单个雌性生殖细胞,用软件拍照存下目标细胞所处的位置信息及编号,然后进行激光切割。将加有细胞收集液的lcm96孔板的板盖置于收集架上,调整好孔的位置,使每个孔在收集细胞时都正对显微镜视野中心,每孔分别收集一细胞。根据组织形貌及细胞位置,若在不同切片层中看到同一细胞,则将细胞的不同部分分别切割,收集至同一样品孔中。收集结束后用自封袋将96孔板密封,立即转入-80℃冰箱。6.细胞裂解将收集有细胞的lcm96孔板(或pcr管)从-80℃冰箱中取出,冰上放置2约0min融化后,4℃,4000rpm离心5min,将细胞收集至管底。在超净台中打开96孔板的盖子70℃敞口加热蒸发至溶液剩余2μl,置于冰盒上。7.rna反转为cdna使用thermoscientific的maximahminusreversetranscriptase(#ep0753)按照实施例1中表2配制反应体系(96孔板量)。每孔加入3μl,即共为5μl反应体系,42℃孵育90分钟。8.cdna纯化及放大将所有pcr管子中的溶液汇总到一个管子中,使用zymoresearch(#d4013)dna纯化试剂盒,最后使用18μl水洗脱cdna,加入2μl10×reactionbuffer和1μlexonucleasei,37℃孵育30分钟,80℃孵育20分钟。使用terrapcrdirectpolymerasemix(clontech,#639270)试剂盒进行cdna放大,按照实施例1中表3配制反应体系。按照实施例1中表4所示的程序进行pcr扩增后4℃保存。使用0.8xbechmanampurexp磁珠纯化扩增后的cdna。9.建库及测序纯化后的cdna应用诺唯赞的trueprepdnalibraryprepkit进行建库,使用p5nextpt5(us14898030a1)作为i5接头,扩增10个循环,使用0.8xbechmanampurexp磁珠纯化后使用安捷伦2100核酸分析仪进行片段分析,文库在300-800bp,后应用illuminanovaseq系统测序。本实施例中的建库流程如图6所示。10.测序数据处理(1)使用cutadapt去除测序数据的接头;(2)使用trimmomatic去除低质量的测序数据;(3)通过细胞uniquebarcode在readsr1中提取出中提取出细胞标签信息;(4)通过r1提取的细胞标签信息对应到r2中,并将r2提取出的各个细胞reads使用hisat2比对到基因参考组上;(5)去除各个细胞下的pcr重复,使用htseq-count对umi进行计数;(6)过滤掉低质量的细胞,具体指标如下滤除umi低于1000、umi高于10000,以及mtrna含量超过10%的细胞。(7)经过数据过滤后,得到单细胞基因表达矩阵。(8)对单细胞数据进行聚类分析,得到主要的雌性生殖细胞类型及其基因表达特征。11.图片信息与测序数据联合分析将lcm分选过程中保存的每一个细胞位置信息的图片利用图像拼接技术进行合并拼接,从而得到卵巢组织的三维组织示图。根据细胞编号将细胞基因表达信息比对回三维的卵巢组织中,从而实现卵巢中每一个生殖细胞的细胞类型、发育阶段、基因表达的注释,从而发现更多新的关于雌性生殖细胞分化命运和其空间位置的关系。讨论与现有的单细胞转录组技术相比,本发明的单细胞空间转录组技术主要有以下优势:1.实现单细胞分辨率的空间转录组分析目前常用的单细胞分选技术如流式细胞分选、口吸管吸取和微流控系统分离均需要将组织预先解离成单细胞悬液,导致细胞在组织中原本的空间位置信息丢失,且较长时间的酶解消化过程,或口吸管产生的环境压力等会对细胞产生强烈刺激,可能使转录组信息发生改变,影响转录组信息的准确性(denisenkoe,guobb,jonesm,etal.genomebiology,2020,21(1);brinks,fsage,belvértesy,etal.naturemethods,2017,14(10).)。而本发明的lcm-scrb-seq方法采用组织原位的激光显微切割,无需消化成细胞悬液可直接在组织切片上进行细胞分选,不仅可以对组织形貌进行直接观察,便于识别记录细胞原有的空间位置,还减少了酶解消化等步骤,尽可能降低了外界酶解及温度的长时间刺激对细胞转录组的影响。2.获得高通量的组织单细胞转录组数据目前有零星报道的,基于激光显微切割技术的组织单细胞转录组方法,都是利用smart技术基于单个细胞的转录组文库构建,通量低,实验工作量很大。本发明的lcm-scrb-seq方法采用96孔的收集板,在建库中对细胞进行barcode标记后,将单细胞的转录本文库混合进行后续操作,大大提高了实验的通量,以及单细胞转录组数据的重复性。3.从组织原位捕获到的单细胞中获得高质量的cdna本发明的lcm-scrb-seq方法进行激光显微切割分选单细胞,mrna反转,cdna扩增并纯化后,得到的cdna进行agilent2100分析质检。质检结果显示(图2),本发明的lcm-scrb-seq方法获得的长度超过1000bp的全长cdna占比超过50%,甚至已超过单细胞转录组金标准smart-seq2所做到的50%左右,满足建库要求。4.灵敏度较高应用本发明的lcm-scrb-seq方法对30天正常小鼠的卵母细胞进行建库测序,在每个细胞1mreads的测序深度下,每个细胞测到的平均基因数为10320(图3),与之前已发表的应用smart-seq对小鼠单个卵母细胞转录组测序测得的基因数基本相当,说明本发明的lcm-scrb-seq方法的灵敏度较高。5.一致性较好将hela细胞同步化到有丝分裂中期,获得基因表达相对均一的细胞,利用本发明的lcm-scrb-seq方法通过激光显微切割将单个贴在玻片上的细胞分选出来建库测序。得到测序结果后将每个细胞中的转录本数进行标准化,并计算单个细胞之间基因表达的相关性。结果发现单个细胞之间基因表达的相关性较高,平均在0.87以上(图4)。表明细胞之间由于方法本身造成的差异较小,本发明的lcm-scrb-seq方法的一致性较好。6.另外,该方法适用于细胞悬液中少量细胞的分选,成为传统分析方法必要的补充目前常用的酶解成单细胞悬液再进行单细胞分选的方法都存在着一定的缺陷,例如流式细胞分选(facs)的细胞量需要达到1×106个,所需细胞初始上样量较大,当样本的细胞数达不到上样量要求时,便无法进行facs分选。而实际应用中,许多珍贵的临床样品可获得的细胞量少,无法利用facs进行分选传统的口吸管法虽然可用于分选少量单细胞,但操作极不便捷,可获得的单细胞通量太小,并且细胞在被吸取的过程中受到强压的刺激,可能会产生应激反应,造成转录本信息的改变。本发明的lcm-scrb-seq方法对入口细胞的大小和数量没有限制,将单细胞悬液涂在带膜玻片上,对细胞的形貌进行具体观察,从而实现对单个细胞的针对性挑选,同时激光切割产生的能量不会对细胞的基因表达产生影响。在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。序列表<110>上海交通大学<120>一种新型组织单细胞空间转录组技术<130>p2020-1189<160>4<170>patentinversion3.5<210>1<211>73<212>dna<213>人工序列(artificialsequence)<220><221>misc_feature<222>(26)..(41)<223>nisa,c,g,ort<220><221>misc_feature<222>(73)..(73)<223>nisa,c,g,ort<400>1acactctttccctacacgacgatctnnnnnnnnnnnnnnnnttttttttttttttttttt60tttttttttttvn73<210>2<211>28<212>dna<213>人工序列(artificialsequence)<400>2cgcacactctttccctacacgacgcggg28<210>3<211>22<212>dna<213>人工序列(artificialsequence)<400>3acactctttccctacacgacgc22<210>4<211>58<212>dna<213>人工序列(artificialsequence)<400>4aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1