一种Tafamidis及其衍生物的合成方法
【技术领域】
本发明涉及有机合成领域,具体涉及一种tafamidis及其衍生物的合成方法。
背景技术:
tafamidis是fda批准的第一种用于治疗由转甲状腺素介导的淀粉样变性病(attr-cm)引起的心肌病药物。该药物由辉瑞公司申请并于2019年在美国获准使用。据临床试验评估,该药物将患者的死亡风险降低了30%,还将药物服用者心血管相关疾病住院率降低了32%。相关的药效学表明,tafamidis可以与甲状腺素转运蛋白(ttr)四聚体的甲状腺素结合位点结合并抑制其解离成单体,从而阻止ttr淀粉样蛋白生成级联反应。
关于tafamidis代表性的合成方法是将4-氨基-3-羟基苯甲酸用二氯苯甲酰氯酰化,所得产物用对甲苯磺酸一水合物处理,生成的残余物用(三甲基甲硅烷基)重氮甲烷酯化,环化,最后水解产生tafamidis。这一过程反应步骤多,总收率较低,所需要的不易得的原料、强酸强碱及添加剂等使该方法的成本大大提高,对环境也造成了极大地压力。【参考文献:(a)sancar,f.j.am.med.assoc.2019,321,2274.321.(b)müller,m.l.;butler,j.;heidecker,b.eur.j.heartfail.2020,22,39.(c)said,g.;grippon,s.;kirkpatrick,p.;naturepublishinggroup:tafamidis.2012.(d)corazza,a.;verona,g.;waudby,c.a.;mangione,p.p.;bingham,r.;uings,i.;canetti,d.;nocerino,p.;taylor,g.w.;pepys,m.b.j.med.chem.2019,62,8274.(e)zhang,y.;ji,m.eur.j.org.chem.2019,2019,7506.(f)kandula,m.2013.wo2013168014.(g)kandula,m.2015.us20150126567.(h)yuan,s.;yu,b.;liu,h.-m.eur.j.med.chem.2020,112667.】
针对上述方法的不足,开发一种以简单原料和简单的反应条件合成tafamidis及其衍生物的有效方法具有重要意义和巨大的应用价值。
技术实现要素:
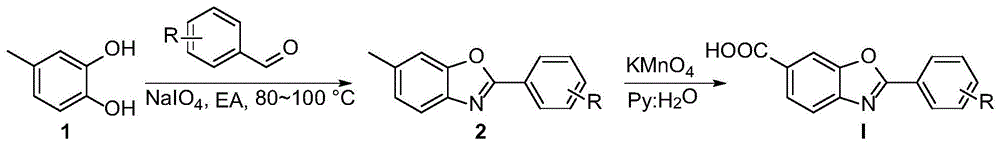
本发明的目的是开发一种以6-氨基间甲酚和醛类化合物为原料,在氧化剂高碘酸钠和高锰酸钾的作用下高转化率地合成tafamidis及其衍生物的方法。
本发明的目的是通过如下技术方案实现的:
一种tafamidis及其衍生物的合成方法,所述tafamidis衍生物的制备原料包括:6-氨基间甲酚、醛类化合物、高碘酸钠(naio4)、高锰酸钾。
所述tafamidis衍生物具有以下结构式(i):
所述结构式中,r为在苯环上的任意一个或多个位置的取代基,且各r独立的选自h、叔丁基、甲氧基、f、cl、br、i、三氟甲基、硝基、甲酸甲酯基。
在一种实施方式中,所述醛类化合物选自苯甲醛、对甲氧基苯甲醛、对叔丁基苯甲醛、对氟基苯甲醛、对氯基苯甲醛、对溴苯甲醛、对碘苯甲醛、3,5-二氯苯甲醛、对三氟甲基苯甲醛、对硝基苯甲醛、对甲酰基苯甲酸甲酯中的一种。
在一种实施方式中,优选的,一种tafamidis及其衍生物的合成方法,包含以下步骤:
(1)取6-氨基间甲酚、醛类化合物1、naio4和溶剂置于反应容器中,混合,加热搅拌反应,反应结束后冷却至室温,用饱和na2co3洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离,即得苯并噁唑类化合物2;
其中,所述6-氨基间甲酚、醛类化合物和naio4之间的摩尔比为1:(1.8~2.2):(1.8~2.2);
优选的,所述6-氨基间甲酚、醛类化合物和naio4之间的摩尔比为1:(1.9~2.1):(1.9~2.1);
更优选的,所述6-氨基间甲酚、醛类化合物和naio4之间的摩尔比为1:2:2;
所述溶剂为n,n-二甲基甲酰胺、1,2-二氯乙烷、乙腈、氯仿、乙酸乙酯中的至少一种;
所述反应的温度为80-100℃;
优选的,所述反应的温度为100℃;
所述反应的时间为3-4h;
优选的,所述反应的时间为3h;
所述惰性气体为氮气、氩气与氦气中的任意一种或多种的组合;
(2)取苯并噁唑类化合物2、kmno4和溶剂置于反应容器中,搅拌,然后加热至沸腾。反应完成后,酸化,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离,即得tafamidis衍生物;
其中,所述苯并噁唑类化合物2和kmno4之间的摩尔比为1:(4~6);
所述溶剂为吡啶和水的混合溶剂;
所述反应的温度为90-100℃;
所述反应的时间为5-6h。
根据实验研究,本发明提供了一种在高碘酸钠和高锰酸钾的作用下由6-氨基间甲酚和醛类化合物制备tafamidis及其衍生物的合成方法。在这一方法中,避免了酰化试剂二氯苯甲酰氯和酯化试剂(三甲基甲硅烷基)重氮甲烷的使用,解决了酰氯、重氮试剂以及强酸强碱添加剂等对环境危害的问题;反应步骤少、总产率较高;甲氧基、卤素、三氟甲基等等都可以被很好地耐受;反应结束后的碘酸盐和二氧化锰不溶于反应溶液,可以被很好地回收,极大地降低了反应体系所带来的污染,为tafamidis及其衍生物的合成提供了一种更加经济环保的路径,具有巨大的应用价值。
【附图简要说明】
图1为制备tafamidis及其衍生物的反应式。
【具体实施方式】
下面结合本发明的合成例对本发明所述的合成方法作进一步说明,需要说明的是,实施例并不构成对本发明要求保护范围的限制。
一种tafamidis及其衍生物的合成方法,所述tafamidis衍生物的制备原料包括:6-氨基间甲酚、醛类化合物、高碘酸钠(naio4)、高锰酸钾。
在一种实施方式中,所述tafamidis衍生物具有以下结构式(i):
在一种实施方式中,所述结构式中,r为在苯环上的任意一个或多个位置的取代基,且各r独立的选自h、叔丁基、甲氧基、f、cl、br、i、三氟甲基、硝基、甲酸甲酯基。
在一种实施方式中,所述醛类化合物选自苯甲醛、对甲氧基苯甲醛、对叔丁基苯甲醛、对氟基苯甲醛、对氯基苯甲醛、对溴苯甲醛、对碘苯甲醛、3,5-二氯苯甲醛、对三氟甲基苯甲醛、对硝基苯甲醛、对甲酰基苯甲酸甲酯中的一种。
在一种实施方式中,优选的,一种tafamidis及其衍生物的合成方法,包含以下步骤:
(1)取6-氨基间甲酚、醛类化合物1、naio4和溶剂置于反应容器中,混合,加热搅拌反应,反应结束后冷却至室温,用饱和na2co3洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离,即得苯并噁唑类化合物2;
在一种实施方式中,所述6-氨基间甲酚、醛类化合物和naio4之间的摩尔比为1:(1.8~2.2):(1.8~2.2);
在一种实施方式中,优选的,所述6-氨基间甲酚、醛类化合物和naio4之间的摩尔比为1:(1.9~2.1):(1.9~2.1);
在一种实施方式中,更优选的,所述6-氨基间甲酚、醛类化合物和naio4之间的摩尔比为1:2:2;
在一种实施方式中,所述溶剂为n,n-二甲基甲酰胺、1,2-二氯乙烷、乙腈、氯仿、乙酸乙酯中的至少一种;
在一种实施方式中,所述反应的温度为80-100℃;
在一种实施方式中,优选的,所述反应的温度为100℃;
在一种实施方式中,所述反应的时间为3-4h;
在一种实施方式中,优选的,所述反应的时间为3h;
在一种实施方式中,所述惰性气体为氮气、氩气与氦气中的任意一种或多种的组合;
(2)取苯并噁唑类化合物2、kmno4和溶剂置于反应容器中,搅拌,然后加热至沸腾。反应完成后,酸化,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离,即得tafamidis衍生物;
在一种实施方式中,所述苯并噁唑类化合物2和kmno4之间的摩尔比为1:(4~6);
在一种实施方式中,所述溶剂为吡啶和水的混合溶剂;
在一种实施方式中,所述反应的温度为90-100℃;
在一种实施方式中,所述反应的时间为5-6h。
根据实验研究,本发明提供了一种在高碘酸钠和高锰酸钾作用下由6-氨基间甲酚和醛类化合物制备tafamidis及其衍生物的合成方法。该方法主要避免了强酸强碱及添加剂的使用,具体表现为:(1)在这一方法中,以廉价易得的6-氨基间甲酚和醛类化合物为原料,经由naio4氧化缩合和高锰酸钾氧化甲基两步合成tafamidis及其衍生物的方法实现,甲氧基、卤素、三氟甲基等等都可以被很好地耐受,(2)避免了酰化试剂二氯苯甲酰氯和酯化试剂(三甲基甲硅烷基)重氮甲烷的使用,(3)解决了酰氯、重氮试剂以及强酸强碱添加剂等对环境危害的问题,(4)反应结束后的碘酸盐和二氧化锰不溶于反应溶液,可以被很好地回收,极大地降低了反应体系所带来的污染,为该新型tafamidis衍生物的合成提供了一种更加经济环保的路径。
下面是具体的合成例。
如图1所示:
合成例1
tafamidis的合成
(1)制备2-(3,5-二氯苯基)-6-甲基苯并[d]噁唑:在反应器中加入1mmol6-氨基间甲酚、2mmol3,5-二氯苯甲醛、2mmolnaio4,10mlea。在氮气氛围下,在100℃条件下持续搅拌5h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率91%。
(2)合成tafamidis:然后将0.4mmol2-(3,5-二氯苯基)-6-甲基苯并[d]噁唑和6eqkmno4添加到吡啶(0.9ml)和水(0.6ml)中的混合溶液中。在室温下将所得溶液搅拌30分钟并加热至沸腾(约100℃)。反应完成后,使用1mhcl溶液将ph调节至ph=2-3,然后用乙酸乙酯和水萃取产物两次。用1mhcl溶液洗涤有机层,去除多余的吡啶。然后用无水硫酸镁(mgso4)干燥有机层并在减压蒸去溶剂。用石油醚/乙酸乙酯在硅胶上柱层析法对粗产物进行纯化,得到相应的tafamidis,收率为65%。
合成例2
2-(4-甲氧基苯基)苯并[d]噁唑-6-羧酸的合成
(1)制备6-甲基-2-(4-甲氧基苯基)苯并[d]噁唑:在反应器中加入1mmol6-氨基间甲酚、2mmol4-甲氧基苯甲醛、2mmolnaio4,10mlea。在氮气氛围下,在100℃条件下持续搅拌5h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率87%。
(2)合成2-(4-甲氧基苯基)苯并[d]噁唑-6-羧酸:然后将0.4mmol6-甲基-2-(4-甲氧基苯基)苯并[d]噁唑和6eqkmno4添加到吡啶(0.9ml)和水(0.6ml)中的混合溶液中。在室温下将所得溶液搅拌30分钟并加热至沸腾(约100℃)。反应完成后,使用1mhcl溶液将ph调节至ph=2-3,然后用乙酸乙酯和水萃取产物两次。用1mhcl溶液洗涤有机层,去除多余的吡啶。然后用无水硫酸镁(mgso4)干燥有机层并在减压蒸去溶剂。用石油醚/乙酸乙酯在硅胶上柱层析法对粗产物进行纯化,得到相应的2-(4-甲氧基苯基)苯并[d]噁唑-6-羧酸,收率为65%。
合成例3
2-(4-溴苯基)苯并[d]噁唑-6-羧酸的合成
(1)制备6-甲基-2-(4-溴苯基)苯并[d]噁唑:在反应器中加入1mmol6-氨基间甲酚、2mmol4-溴苯甲醛、2mmolnaio4,10mlea。在氮气氛围下,在100℃条件下持续搅拌5h,停止反应,冷却至室温,用饱和nacl洗涤,然后用乙酸乙酯萃取,减压蒸馏浓缩除去溶剂,干燥,粗产品经柱色谱分离即得目标产物,产率80%。
(2)合成2-(4-溴苯基)苯并[d]噁唑-6-羧酸:然后将0.4mmol6-甲基-2-(4-溴苯基)苯并[d]噁唑和6eqkmno4添加到吡啶(0.9ml)和水(0.6ml)中的混合溶液中。在室温下将所得溶液搅拌30分钟并加热至沸腾(约100℃)。反应完成后,使用1mhcl溶液将ph调节至ph=2-3,然后用乙酸乙酯和水萃取产物两次。用1mhcl溶液洗涤有机层,去除多余的吡啶。然后用无水硫酸镁(mgso4)干燥有机层并在减压蒸去溶剂。用石油醚/乙酸乙酯在硅胶上柱层析法对粗产物进行纯化,得到相应的2-(4-溴苯基)苯并[d]噁唑-6-羧酸,收率为56%。
- 还没有人留言评论。精彩留言会获得点赞!