三重数字微滴PCR检测猪圆环病毒的方法、引物、探针和试剂盒与流程
本发明涉及防疫检测领域,尤其涉及一种三重数字微滴pcr检测猪圆环病毒的方法、引物、探针和试剂盒。
背景技术:
根据最新的oie发布的《陆生动物卫生法典》,饲料、肉制品等动物源深加工产品被定义为传播动物疫病的重要风险点。猪圆环病毒作为二类传染疫病,主要分为猪圆环病毒1型、猪圆环病毒2型、猪圆环病毒3型,在全世界的养猪场里广泛传播。由于我国猪肉及其产品消费需求广泛,加上pcv2自身耐高温耐酸等特性,使得猪源性产制品成为传播pcv2的重要生物安全风险点。
实时荧光定量pcr(real-timefluorescentquantitativepcr,qpcr)作为目前应用最为广泛的核酸检测方法,通过标准曲线可对样品中的病毒核酸进行检测定量。qpcr法在检测动物组织等常规样品时具有一定的优势,但动物源产制品,尤其是饲料、肉制品等深加工产品往往成分基质复杂,且含有大量抑制物,使得qpcr等常规方法受干扰程度较大,容易出现假阴性结果。数字微滴pcr(digitaldropletpcr,ddpcr)通过乳化、微孔板、列阵芯片等方式获得大量的独立微小反应体系,使抑制剂以及其他杂质对扩增效率的影响大大削弱,提高了检测的稳定性和灵敏度,甚至可不处理样品直接进行ddpcr因其高灵敏度。强抗干扰性以及绝对定量等优点使得ddpcr近年来在艾滋病毒、乙型肝炎病毒、寨卡病毒及新型冠状病毒等人类病毒载量滴定,以及猪繁殖与呼吸综合征病毒、狂犬病毒等动物病毒的检测中已被证明具有很高的灵敏度以及特异性。
技术实现要素:
为了解决上述的技术问题,本发明基于ddpcr的优势,建立猪圆环病毒不同基因型的高灵敏、精确鉴别检测方法,可应用于常规动物检疫以及饲料、肉制品、疫苗等病毒含量低、基质复杂的动物源产制品的检疫监测和载量滴定。
本发明的一个目的是提供检测靶点在检测猪圆环病毒中的应用,所述的检测为非疾病的检测与诊断方法,检测靶点包括以下检测靶点中的一种或多种:
猪圆环病毒1型(pcv1)检测靶点的序列如seqidno:1所示;
猪圆环病毒2型(pcv2)检测靶点的序列如seqidno:2所示;
猪圆环病毒3型(pcv3)检测靶点的序列如seqidno:3所示。
进一步,本发明还提供了一种检测猪圆环病毒的检测靶点的引物,检测靶点包括以下检测靶点中的一种或多种:
pcv1的检测靶点的序列如seqidno:1所示;
pcv2的检测靶点的序列如seqidno:2所示;
pcv3的检测靶点的序列如seqidno:3所示。
优选,本发明的引物包括以下的一种或多种:
pcv1的检测靶点的引物
fp1:ggtgtggcgggaggagtag
rp1:ttgggtccactgttgttat;
pcv2的检测靶点的引物
fp2:gaaatggtatttgggtgccc
rp2:actccgttgtccttgagatc;
pcv3的检测靶点的引物
fp3:gtgccgtagaagtctgtcattc
rp3:caggacaaagcctcttcttttt。
进一步,本发明还提供了一种检测猪圆环病毒的检测靶点的探针,检测靶点包括以下检测靶点中的一种或多种:
pcv1的检测靶点的序列如seqidno:1所示;
pcv2的检测靶点的序列如seqidno:2所示;
pcv3的检测靶点的序列如seqidno:3所示。
优选,探针包括以下的一种或多种:
pcv1的检测靶点的探针p1:ccaagttggtggagggggtt;
pcv2的检测靶点的探针p2:agcgaaaggaacagatcagc
pcv3的检测靶点的探针p3:ataaatgctccaaagcagtgctcc。
优选,pcv3的探针p3以hex作为荧光发光基团,pcv2探针标记fam荧光基团,pcv1探针p1以cy5作为发光基团。
进一步,本发明还提供了一种试剂盒,该试剂盒包括:
所述的引物;和/或,所述的探针。
进一步,本发明还提供了一种三重数字微滴pcr检测猪圆环病毒的方法,该方法为非疾病的检测与诊断方法,该方法包括以下的步骤:配制pcr反应体系、芯片上样、微滴生成、扩增反应和微滴信号读取与分析;pcr反应体系的引物采用所述的引物,芯片所述的探针。
优选,该方法包括以下的步骤:
1)反应体系配制
cdpcr采用上述提过的引物和探针,配制成25μl体系;使用涡旋仪将配好的pcr反应液进行充分混匀,再通过离心机去除气泡。
2)芯片上样
2.1)在取出一片新的sapphire芯片,打开芯片的白色盖子,任意方向旋转1/4圈即可打开,并丢弃;
2.2)移取25μl混合好的pcr反应液,加入到sapphire芯片,注意吸头不要取内置油相接触,注意不能产生气泡;
2.3)利用专用pcr盖封闭孔井;
3)微滴生成与pcr扩增
3.1)将封闭好的芯片转移到naicageode微滴生成和扩增系统中,关上仓门,按照给定程序进行微滴发生;
3.2)发生结束后自动进入pcr反应;
4)微滴信号读取和分析
将完成pcr反应的芯片取出,小心缓慢水平转移到prism3系统中;选择好荧光通道和芯片位置后开始读取芯片中的微滴数和荧光值;仪器会自动对每个芯片拍照,并对芯片的中的每个微滴进行三色检测器扫描,确定阴阳性微滴信号;然后根据泊松分布原理计算出绝对检测浓度。
再优选,本发明cdpcr退火温度为59℃,上下游引物终浓度为0.5μmol/l;探针浓度为0.5μmol/l。
再优选,本发明cdpcr反应体系为25μl:5×cdpcrsupermix5μl,fp1(20μmol/l)0.625μl,rp1(20μmol/l)0.625μl,p1(20μmol/l)0.625μl,fp2(20μmol/l)0.625μl,rp2(20μmol/l)0.625μl,p2(20μmol/l)0.625μl,fp3(20μmol/l)0.625μl,rp3(20μmol/l)0.625μl,p3(20μmol/l)0.625μl,模板1μl,h2o13.375μl。
再优选,本发明cdpcr扩增步骤如下:
再优选,本发明所有阴性对照的荧光值确定基线阈值:cy5通道的荧光阈值为10729、fam通道的荧光阈值为17542以及hex通道为10729。
本发明由于采用了上述的技术方案,根据pcv1基因序列(kc447455.1)、pcv2基因序列(mg245866)和pcv3基因序列(nc_031753.1)为模板,分别设计对应的特异性引物及探针,在进行pcr退火温度优化、qpcr引物终浓度优化以及三重crystal数字pcr探针终浓度优化反应后,建立了三重crystal数字pcr检测pcv1、pcv2、pcv3的方法。同时也通过qpcr方法来进行比较。建立的标准曲线结果显示,三重crystal数字pcr在检测pcv1、pcv2、pcv3均具有良好的线性关系,它们的相关系数分别为:rpcv12=0.9878、rpcv22=0.9966、rpcv32=0.9833,扩增效率分别为:epcv1=90%、epcv2=98%、epcv3=94%,表明其具有良好的扩增效率和相关性;该法的特异性良好,只能特异性扩增相关基因,建立的三重crystal数字pcr最低能够检测到质粒标准品的拷贝数为pcv1:4.96拷贝/μl、pcv2:12.29拷贝/μl、pcv3:11.54拷贝/μl。抗干扰试验表明该法的最低检测限不受到相似病毒的影响。且重复性试验表明,组内重复性变异系数均在10%内,组间变异系数在106拷贝/μl~101拷贝/μl数量级时变异系数良好,在100拷贝/μl数量级时较为一般,但均能检出。说明建立的三重crystaldpcr具有良好的重复性,具有市场应用潜力。
附图说明
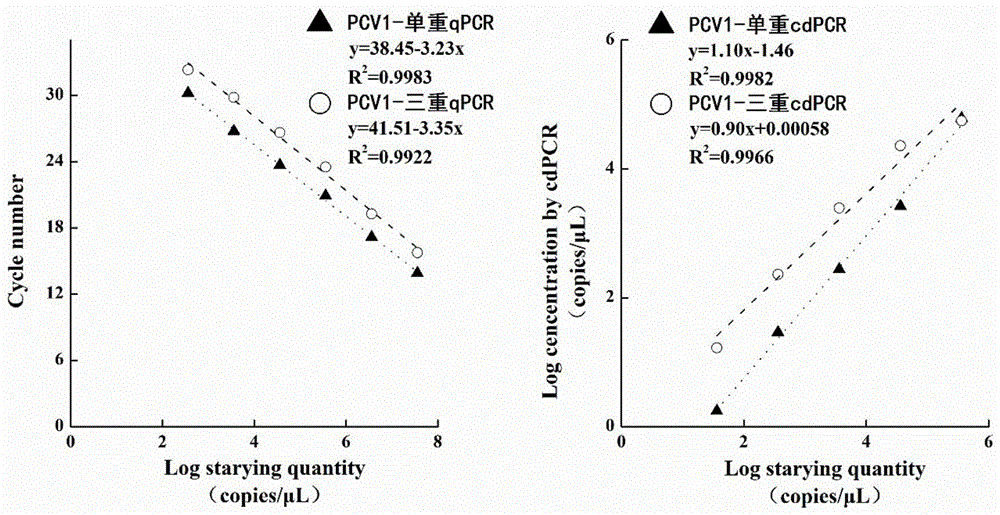
图1为pcv1标准曲线。
图2为pcv2标准曲线。
图3为pcv3标准曲线。
图4为三重crystaldpcr体系特异性试验结果图。
图5为dpcr敏感性试验图。
图6为三重cdpcr体系下高中低浓度的pcv2和pcv3对pcv1的干扰性分析图。
图7为三重cdpcr体系下高中低浓度的pcv1和pcv3对pcv2的干扰性分析图。
图8为三重cdpcr体系下高中低浓度的pcv1和pcv2对pcv3的干扰性分析图。
图9为pcv1(a)、pcv2(b)和pcv3(c)三重cdpcr反应体系批间重复性试验图。
具体实施方式
一、引物和探针的设计与合成
根据genbank中登录的pcv1基因序列(kc447455.1)、pcv2基因序列(mg245866)和pcv3基因序列(nc_031753.1)为模板,并利用primer3分别针对pcv1、pcv2和pcv3设计特异性引物和特异性taqman探针,其中pcv3的探针p3以hex作为荧光发光基团,pcv2探针标记fam荧光基团,pcv1探针p1以cy5作为发光基团(表1)。并根据各个基因组序列来设计质粒标准品。引物和探针由takara公司合成。
表1扩增引物及探针序列
二、dpcr检测方法的建立
2.1dpcr的工作流程
采用naicatmcrystaldigitalpcr系统进行crystaldpcr(以下简称cdpcr)扩增,实验操作步骤包括配制pcr反应体系、芯片上样、微滴生成、扩增反应、微滴信号读取与分析。
(1)反应体系配制
cdpcr采用上述提过的引物和探针,配制成25μl体系。使用涡旋仪将配好的pcr反应液进行充分混匀,再通过离心机去除气泡。
(2)芯片上样
①在取出一片新的sapphire芯片,打开芯片的白色盖子,任意方向旋转1/4圈即可打开,并丢弃;
②移取25μl混合好的pcr反应液,加入到sapphire芯片,注意吸头不要取内置油相接触,注意不能产生气泡;
③利用专用pcr盖封闭孔井。
(3)微滴生成与pcr扩增
①将封闭好的芯片转移到naicageode微滴生成和扩增系统中,关上仓门,按照给定程序进行微滴发生;
②发生结束后自动进入pcr反应。
(4)微滴信号读取和分析
将完成pcr反应的芯片取出,小心缓慢水平转移到prism3系统中。选择好荧光通道和芯片位置后开始读取芯片中的微滴数和荧光值。仪器会自动对每个芯片拍照,并对芯片的中的每个微滴进行三色检测器扫描,确定阴阳性微滴信号。然后根据泊松分布原理计算出绝对检测浓度。
2.2dpcr反应条件的优化
(1)退火温度、引物探针浓度的优化
依旧使用t100thermalcyclerpcr扩增仪来设定6组不同的退火温度(53℃、55℃、57℃、59℃、61℃)进行普通pcr,反应结束后进行跑胶,根据条带亮度和清晰度确定最佳退火温度。以标准质粒为模板,在一样的qpcr反应体系中,设置不同浓度的上下游引物:0.5μmol/l、0.7μmol/l、0.9μmol/l,根据ct值和扩增效率确定最佳引物浓度。以标准质粒为模板,在相同的qpcr反应体系里,设置不同探针终浓度:分别为0.25μmol/l和0.5μmol/l进行试验,通过阴阳性微滴分布情况和阳性微滴数来确定最佳探针浓度。
(2)荧光阈值的优化
根据最优反应体系和最佳退火温度后,进行12个阴性对照cdpcr反应,获得本方法的荧光阈值,从而确定后续样品的实际拷贝数。
2.3标准曲线的建立及对比
优化试验结束后,将pcv1、pcv2、pcv3质粒标准品分别进行连续10倍梯度稀释,分别获得理论浓度3.61×107拷贝/μl~3.61拷贝/μl、3.23×106拷贝/μl~3.23拷贝/μl和3.16×106拷贝/μl~3.16拷贝/μl共22个系列。以它们为模板分别进行三重cdpcr、单重cdpcr、三重qpcr和单重qpcr,分别获得反应结束后的实际检测拷贝数和ct值,使用origin8软件计算、分析并绘制出这两种方法的标准曲线同时获得扩增效率。
2.4特异性试验
分别提取猪繁殖与呼吸道综合征病毒、伪狂犬病病毒、猪圆环病毒1型、2型和3型、猪瘟病毒、非洲猪瘟病毒的核酸,以这些病毒的核酸为模板,进行三重cdpcr反应,以确定该方法的特异性。
2.5敏感性试验
按10倍倍比稀释不同类型的pcv质粒标准品,分别获得pcv1理论浓度3.61×107拷贝/μl~3.61拷贝/μl共8个系列、pcv2理论浓度3.23×106拷贝/μl~3.23拷贝/μl共7个系列和pcv3理论浓度3.16×106拷贝/μl~3.16拷贝/μl共7个系列。理论浓度每个模板浓度重复6次,同时将该反应体系应用到qpcr当中,分析两个方法各自的敏感性。
同时,将不同型pcv阳性dna按10倍倍比稀释后1:1:1混合,按照棋盘滴定方法进行检测,用以分析多重cdpcr体系在不同dna模板存在的情况下对另一型pcvdna的最低检测限的影响。
2.6重复性试验
将不同基因型pcv质粒标准品按10倍倍比稀释,每个稀释度设置6个重复进行实验,计算各个稀释度的平均浓度和标准差,来评估该法的批内重复性,每次试验均设立阴性对照。同时将上述质粒标准品10倍倍比稀释后分别经4次不同时间检测,每个稀释度设置3个重复,计算4批次独立试验的各稀释度的平均浓度以及标准差,评估该方法的批间重复性,每次试验均设立阴性对照即不含样品的25μlcdpcr反应体系。
三、结果
3.1退火温度的优化
根据普通pcr的实验结果,最终确定了59℃为cdpcr的最佳退火温度,该实验pcr扩增体系如表2所示
表2crystaldpcr扩增体系
*ap为室压。
3.2引物浓度的优化
根据qpcr优化结果,不管是0.5μmol/l、0.7μmol/l还是0.9μmol/l,它们之间的扩增效率都是理想状态,因此选择上下游引物终浓度为0.5μmol/l。
3.3探针浓度的优化
根据cdpcr阴阳微滴分布情况最后确定了0.5μmol/l为本研究最佳的探针浓度。因此,本方法最终确定cdpcr反应体系为25μl:5×cdpcrsupermix5μl,fp1(20μmol/l)0.625μl,rp1(20μmol/l)0.625μl,p1(20μmol/l)0.625μl,fp2(20μmol/l)0.625μl,rp2(20μmol/l)0.625μl,p2(20μmol/l)0.625μl,fp3(20μmol/l)0.625μl,rp3(20μmol/l)0.625μl,p3(20μmol/l)0.625μl,模板1μl,h2o13.375μl。
3.4阈值的优化
按照优化后的反应条件对阴性对照进行12次的重复试验,通过划定所有阴性对照的荧光值确定基线阈值,确定cy5通道的荧光阈值为10729、fam通道的荧光阈值为17542以及hex通道为10729。由于naicatmcrystal全自动微滴芯片数字pcr仪系统可以追溯到每块芯片上每个微滴的发光情况,实际试验过程的阈值主要还是以系统自动生成为主,再根据中间雨滴发光情况确定最后各个通道的阈值。
3.5crystaldpcr与qpcr标准曲线的建立及对比
连续10倍梯度稀释pcv1、pcv2、pcv3各自质粒标准品,分别获得理论浓度3.61×107拷贝/μl~3.61拷贝/μl、3.23×106拷贝/μl~3.23拷贝/μl和3.16×106拷贝/μl~3.16拷贝/μl共22个系列。按照上述优化后的反应条件进行分别进行单重cdpcr、三重cdpcr、单重qpcr和三重qpcr,每个浓度设6个平行重复,并设立阴性对照,根据平均值进行线性回归。将相关数据汇总到表4中,并绘制相应的标准曲线图(图1、图2和图3)。跟据表3可以得出pcv1、pcv2和pcv3无论是单重条件下的cdpcr还是三重条件下的cdpcr,它们的扩增效率都处于理想状态,与传统qpcr不论是单重还是三重结果相比都相差不大,特别是在三重体系下检测pcv3,cdpcr的扩增效率明显比qpcr效果好。并且不同型的pcv的扩增效率在单重和三重的条件下均能保持一致,证明了实验的可靠性和准确性。
表3pcvcdpcr和qpcr试验的扩增效率、相关系数和曲线斜率汇总
3.6特异性试验
本实验建立的三重cdpcr方法对prrsv、csfv、asfv和prv常见猪病病原进行检测,结果显示fam通道只有pcv2有阳性微滴,其他样品没有阳性信号;cy5通道只有pcv1有阳性微滴,其他病毒没有信号;hex通道只有pcv3有阳性微滴,而其他病原均无阳性信号(图4),表明该方法的特异性较强。
3.7敏感性试验
连续10倍梯度稀释pcv1、pcv2、pcv3各自的质粒标准品,分别获得理论浓度3.61×107拷贝/μl~3.61拷贝/μl、3.23×106拷贝/μl~3.23拷贝/μl和3.16×106拷贝/μl~3.16拷贝/μl共22个系列。分别进行三重cdpcr和三重qpcr反应,cdpcr反应循环数设定为45个循环,qpcr反应循环数设定为40个循环。每个浓度设6个平行重复,同时设立3个阴性对照,反应结束后对6个重复样品的平均值进行敏感性计算与分析。
敏感性试验结果显示,三重cdpcr对pcv1质粒标准品的最低检测限为4.96拷贝/μl(表4),质粒标准品理论浓度为3.61×107拷贝/μl时显示体系中所有反应微滴均为阳性信号(图5),说明已超过cdpcr方法的检测浓度上限,表明建立的三重cdpcr体系的检测pcv1范围为5.59×104拷贝/μl~4.96拷贝/μl。而qpcr的最低检测限约为16.75拷贝/μl(表4)。
三重cdpcr对pcv2质粒标准品的最低检测限为12.29拷贝/μl(表4),质粒标准品理论浓度为3.23×106拷贝/μl时显示体系中所有反应微滴均为阳性信号(图5),说明已超过cdpcr方法的检测浓度上限,表明建立的三重cdpcr体系的检测pcv2范围为4.93×104拷贝/μl~12.29拷贝/μl。而qpcr的最低检测限约为57拷贝/μl(表4)。
三重cdpcr对pcv3质粒标准品的最低检测限为11.54拷贝/μl(表4),质粒标准品理论浓度为3.16×106拷贝/μl时显示体系中所有反应微滴均为阳性信号(图5),说明已超过cdpcr方法的检测浓度上限,表明建立的三重cdpcr体系的检测pcv3范围为5.06×104拷贝/μl~11.54拷贝/μl。而qpcr的最低检测限约为33.87拷贝/μl(表4)。
上述三个结果表明三重cdpcr在检测pcv1方面灵敏度比qpcr高出4倍,在检测pcv2时灵敏度高出4倍,在检测pcv3方面比qpcr灵敏度高出2倍。该三重检测pcv方法的敏感性较强,均比传统的qpcr方法灵敏,可以在实际应用方面与qpcr进行互补。
表4三重crystaldpcr体系下pcv1、pcv2、pcv3各自的敏感性及批内重复情况
*体系中的反应微滴均显示阳性,表明超过ddpcr检测范围。
**未检出。为了进一步分析样本中三种不同型的pcv基因组相互干扰情况,将pcv1、pcv2和pcv3质粒标准品按10倍梯度连续稀释后,1:1:1混合成27个混合样,这27个混合样涵盖了pcv1、pcv2和pcv3拷贝数从高浓度到低浓度的情况。结果显示,无论是高浓度(106拷贝/μl数量级)还是低浓度(10拷贝/μl数量级)的pcv2基因组(图6y轴)和pcv3基因组均不影响pcv1质粒标准品的检测(图6z轴),尤其最低浓度梯度(实际平均值为4.96拷贝/μl)pcv1质粒标准品未受pcv2和pcv3基因组存在的影响,所有重复均稳定扩增,表明ddpcr反应体系中pcv1的高效扩增反应不受其他pcv基因组存在的干扰。对pcv2干扰试验的结果与pcv1类似,无论是高浓度(106拷贝/μl数量级)还是低浓度(10拷贝/μl数量级)的pcv1基因组(图7y轴)和pcv3基因组均不影响pcv2质粒标准品的检出,并且最低浓度梯度也未收到影响,表明ddpcr反应体系中pcv2的高效扩增反应不受其他pcv基因组存在的干扰。在对pcv3干扰性实验中,无论是高浓度(106拷贝/μl数量级)还是低浓度(10拷贝/μl数量级)的pcv1基因组(图8y轴)和pcv2基因组均不影响pcv3质粒标准品的检出,并且最低浓度梯度也未收到影响,表明ddpcr反应体系中pcv3的高效扩增反应不受其他pcv基因组存在的干扰。
3.8重复性试验
将10倍连续稀释的pcv1(3.61×107拷贝/μl~3.61拷贝/μl)、pcv2(3.23×106拷贝/μl~3.23拷贝/μl)和pcv3(3.16×106拷贝/μl~3.16拷贝/μl)质粒标准品进行三重cdpcr试验,每个浓度梯度都要进行4次不同时间重复,每次试验时各浓度设3个平行重复,最终获得批间重复结果(图9)。同时也将10倍连续稀释的pcv1(3.61×107拷贝/μl~3.61拷贝/μl)、pcv2(3.23×106拷贝/μl~3.23拷贝/μl)和pcv3(3.16×106拷贝/μl~3.16拷贝/μl)质粒标准品进行批内重复试验,每个浓度做6个平行。实验结果表明,pcv1的批内变异系数为1.08%~6.87%;pcv2的批内变异系数为0.33%~6.99%;pcv3的批内变异系数为0.45%~8.70%。表明该三重cdpcr的检测方法重复性较好,并且批间重复性试验结果证明,三重cdpcr体系在理想浓度为102~105拷贝/μl数量级上批间重复性较好,无显著差异,在理想浓度101拷贝/μl数量级上有差异性但均能检出。
3.9实际样品检测应用
分别利用本研究建立的ddpcr体系和商品化qpcr检测试剂盒分别对组织样品和宠物饲料样品进行检测。结果显示对于猪的组织样本(n=46),qpcr和ddpcr对于不同基因型pcv的检测结果一致,均分别检出pcv1、pcv2和pcv3核酸阳性17、30和8份,其余样本均为阴性。但对于饲料样品中pcv的检测,ddpcr检出pcv2(n=14)和pcv3(n=2)核酸阳性样本数均高于qpcr(表5),ddpcr检测阳性而qpcr检测为阴性的样本中,其pcv核酸拷贝数在7.6-32拷贝/μl,结果表明ddpcr对于复杂基质样本的检测比qpcr具有更高的敏感性和抗干扰性。
表5样品中pcv的ddpcr和qpcr检测
以上为对本发明实施例的描述,通过对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的。本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施列,而是要符合与本文所公开的原理和新颖点相一致的最宽的范围。
序列表
<110>浙江省检验检疫科学技术研究院
<120>三重数字微滴pcr检测猪圆环病毒的方法、引物、探针和试剂盒
<160>12
<170>siposequencelisting1.0
<210>1
<211>95
<212>dna
<213>猪圆环病毒1型(pcv-1)
<400>1
ggtgtggcgggaggagtagttaatataggggtcataggccaagttggtggagggggttac60
aaagttggcatccaagataacaacagtggacccaa95
<210>2
<211>133
<212>dna
<213>猪圆环病毒2型(pcv-2)
<400>2
cccggaaaccacatactggaaaccacctagaaacaagtggtgggatggttaccatggtga60
agaagtggttgttattgatgacttttatggctggctgccgtgggatgatctactgagact120
gtgtgatcgatat133
<210>3
<211>133
<212>dna
<213>猪圆环病毒3型(pcv-3)
<400>3
gtgccgtagaagtctgtcattccagttttttccgggacataaatgctccaaagcagtgct60
ccccattgaacggtggggtcatatgtgttgagccatggggtgggtctggagaaaaagaag120
aggctttgtcctg133
<210>4
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>4
ggtgtggcgggaggagtag19
<210>5
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>5
ttgggtccactgttgttat19
<210>6
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>6
ccaagttggtggagggggtt20
<210>7
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>7
gaaatggtatttgggtgccc20
<210>8
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>8
actccgttgtccttgagatc20
<210>9
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>9
agcgaaaggaacagatcagc20
<210>10
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>10
gtgccgtagaagtctgtcattc22
<210>11
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>11
caggacaaagcctcttcttttt22
<210>12
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>12
ataaatgctccaaagcagtgctcc24
- 还没有人留言评论。精彩留言会获得点赞!