一种新型HuR抑制剂1-苯磺酰基-3-苯基吲哚-4,5-二酮的合成方法
本发明属于有机合成技术领域,具体涉及一种新型hur抑制剂1-苯磺酰基-3-苯基吲哚-4,5-二酮的合成方法。
背景技术:
人类抗原r(hur)是一种在机体组织广泛表达的rna结合蛋白,作为基因转录后表达的重要调控因子;hur调节靶mrna的表达,编码参与炎症,细胞分裂,肿瘤发生,血管生成,免疫等。有研究发现1-苯磺酰基-3-苯基吲哚-4,5-二酮,即化合物5能直接与hur蛋白结合,并调节其与细胞内靶mrna的结合,是一种有效的hur抑制剂。目前,1-苯磺酰基-3-苯基吲哚-4,5-二酮的合成方法报道的较少,以5-甲氧基吲哚为起始原料,3位溴化后,1位氮与苯磺酰基缩合,再与苯基硼酸发生suzuki偶联反应,得到1-苯磺酰基-3-苯基-5-甲氧基吲哚,最后经去甲基化和用ibx氧化得到1-苯磺酰基-3-苯基吲哚-4,5-二酮。该合成方法合成路线较长,操作复杂,中间体3-溴-5-甲氧基吲哚性质不稳定而且反应时间较长,所用的液溴有着强烈的毒害性和腐蚀性,健康成本较高。因此,如何高效,简单的合成1-苯磺酰基-3-苯基吲哚-4,5-二酮对于hur抑制剂的发现和优化至关重要。
技术实现要素:
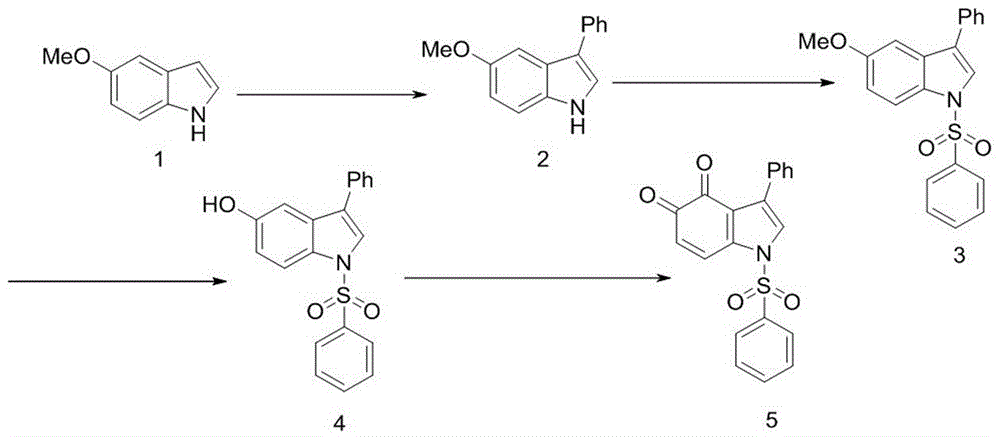
本发明的目的在于提供一种新型hur抑制剂1-苯磺酰基-3-苯基吲哚-4,5-二酮的合成方法,具体步骤为:以5-甲氧基吲哚1为起始原料,在c3与芳基卤化物区域选择性芳基化得到化合物3-苯基-5-甲氧基吲哚2,并用苯磺酰氯处理得到化合物3,经去甲基化得到化合物4,经ibx氧化得到目标化合物1-苯磺酰基-3-苯基吲哚-4,5-二酮5;具体合成路线见图1。
化合物3-苯基-5-甲氧基吲哚2的制备过程为:在干燥的圆底瓶中加入5-甲氧基吲哚和叔丁醇钾,固体混合物经过三次真空/氮气吹扫,然后注入脱气的二甲基亚砜,搅拌使固体溶解,然后滴加碘苯,80℃氮气气氛下搅拌;反应40h后,加入去离子水淬灭,乙酸乙酯萃取,然后依次用半饱和氯化钠溶液,饱和氯化钠溶液洗涤3次,有机层用无水硫酸钠干燥;过滤,滤液减压蒸除溶剂,残余物经硅胶柱层析纯化得到化合物2。
5-甲氧基吲哚,碘苯和叔丁醇钾的投料摩尔比为2:4:1,80℃加热反应。
化合物3的制备过程为:取氢氧化钠和相转移催化剂分散在干燥的二氯甲烷中,加入3-苯基-5-甲氧基吲哚2室温搅拌2min,在冰水浴条件下加入苯磺酰氯,室温反应,2h后薄层色谱检测反应完全,用硅藻土过滤,滤液减压蒸除溶剂,残余物经硅胶柱层析得到化合物3。
相转移催化剂为四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵。
去甲基化的反应过程为:取化合物3,加入干燥的二氯甲烷2.5ml,在-78℃和氮气保护的条件下,加入三溴化硼;将反应置于-78℃的条件下反应0.5h后,将温度逐渐升至室温并用薄层色谱检测反应的情况,2.5h后薄层色谱检测反应完全;向反应液中加入48ml水稀释反应液,用饱和碳酸氢钠溶液中和反应液,用二氯甲烷萃取有机层,有机层用饱和氯化钠溶液洗并用无水硫酸钠干燥,过滤,滤液减压蒸除溶剂,残余物经硅胶柱层析纯化得到化合物4。
ibx氧化的反应过程为:取化合物4溶于溶剂中,然后加入2-碘酰基苯甲酸,室温反应过夜;薄层色谱检测反应完后用硅藻土过滤,滤液减压蒸除溶剂,经硅胶柱层析的到化合物5。
所述的溶剂为dmf或乙酸乙酯。
本发明优点:
目前,1-苯磺酰基-3-苯基吲哚-4,5-二酮的合成方法报道的较少,与现有合成方法相比,此方法具有合成方法新颖,成本低廉,操作简单等优点。1-苯磺酰基-3-苯基吲哚-4,5-二酮是一种有效的hur抑制剂和潜在的抗肿瘤药物靶点。该合成路线的设计为1-苯磺酰基-3-苯基吲哚-4,5-二酮研究体内活性抗癌化合物提供了保障并为开发和研究下一代hur抑制剂奠定了基础。
附图说明
图1为本发明的合成路线;
图2为本发明所述的化合物2的核磁共振氢谱(1hnmr)图;
图3为本发明所述的化合物3的核磁共振氢谱(1hnmr)图;
图4为本发明所述的化合物4的核磁共振氢谱(1hnmr)图;
图5为本发明所述的化合物5的核磁共振氢谱(1hnmr)图。
具体实施方式
下面对本发明作进一步的说明,但不以任何方式对本发明加以限制,基于本发明所作的任何变换,均属于本发明的保护范围。
一种新型hur抑制剂1-苯磺酰基-3-苯基吲哚-4,5-二酮的合成方法,具体步骤为:以5-甲氧基吲哚1为起始原料,在c3与芳基卤化物区域选择性芳基化得到化合物3-苯基-5-甲氧基吲哚2,并用苯磺酰氯处理得到化合物3,经去甲基化得到化合物4,经ibx氧化得到目标化合物1-苯磺酰基-3-苯基吲哚-4,5-二酮5;具体合成路线见图1。
化合物3-苯基-5-甲氧基吲哚2的制备过程为:在干燥的圆底瓶中加入5-甲氧基吲哚和叔丁醇钾,固体混合物经过三次真空/氮气吹扫,然后注入脱气的二甲基亚砜,搅拌使固体溶解,然后滴加碘苯,80℃氮气气氛下搅拌;反应40h后,加入去离子水淬灭,乙酸乙酯萃取,然后依次用半饱和氯化钠溶液,饱和氯化钠溶液洗涤3次,有机层用无水硫酸钠干燥;过滤,滤液减压蒸除溶剂,残余物经硅胶柱层析纯化得到化合物2。
5-甲氧基吲哚,碘苯和叔丁醇钾的投料摩尔比为2:4:1,80℃加热反应。
化合物3的制备过程为:取氢氧化钠和相转移催化剂分散在干燥的二氯甲烷中,加入3-苯基-5-甲氧基吲哚2室温搅拌2min,在冰水浴条件下加入苯磺酰氯,室温反应,2h后薄层色谱检测反应完全,用硅藻土过滤,滤液减压蒸除溶剂,残余物经硅胶柱层析得到化合物3。
相转移催化剂为四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵。
去甲基化的反应过程为:取化合物3,加入干燥的二氯甲烷2.5ml,在-78℃和氮气保护的条件下,加入三溴化硼;将反应置于-78℃的条件下反应0.5h后,将温度逐渐升至室温并用薄层色谱检测反应的情况,2.5h后薄层色谱检测反应完全;向反应液中加入48ml水稀释反应液,用饱和碳酸氢钠溶液中和反应液,用二氯甲烷萃取有机层,有机层用饱和氯化钠溶液洗并用无水硫酸钠干燥,过滤,滤液减压蒸除溶剂,残余物经硅胶柱层析纯化得到化合物4。
ibx氧化的反应过程为:取化合物4溶于溶剂中,然后加入2-碘酰基苯甲酸,室温反应过夜;薄层色谱检测反应完后用硅藻土过滤,滤液减压蒸除溶剂,经硅胶柱层析的到化合物5。
所述的溶剂为dmf或乙酸乙酯。
下面以具体实施案例对本发明做进一步说明:
实施例1
苯基-5-甲氧基吲哚(2)的合成
在干燥的圆底瓶中加入5-甲氧基吲哚(3.0mmol,442.0mg)和叔丁醇钾(6.0mmol,694.0mg),固体混合物经过三次真空/氮气吹扫,然后注入6.5ml脱气的二甲基亚砜(冷冻-泵-解冻),搅拌5min后使固体溶解,然后滴加碘苯(1.5mmol,171ul),80℃氮气气氛下搅拌。反应40h后,加入6ml去离子水淬灭,60ml乙酸乙酯萃取,然后依次用半饱和氯化钠溶液,饱和氯化钠溶液洗涤3次,有机层用无水硫酸钠干燥。过滤,滤液减压蒸除溶剂,残余物经硅胶柱层析纯化得到化合物2162.2mg(产率24%,37%brsm),回收原料5-甲氧基吲哚153.5mg。
4-苯基-5-甲氧基吲哚(2):淡黄色油状液体,v(石油醚):v(乙酸乙酯)=5:1,rf:0.36。1hnmr(cdcl3,400mhz),δ:8.14(s,1h),7.68–7.64(m,2h),7.46(t,j=7.7hz,2h),7.40(d,j=2.3hz,1h),7.35–7.29(m,3h),6.93(dd,j=8.8,2.4hz,1h),3.88(s,3h).13c13nmr(101mhz,cdcl3)δ154.77,135.70,131.84,128.78,127.38,126.21,125.90,122.56,118.21,112.70,112.06,101.70,56.01.核磁共振氢谱(1hnmr)见图2。
1-苯磺酰基-3-苯基-5-甲氧基吲哚(3)的合成
取氢氧化钠(1.86mmol,78mg)和四丁基溴化铵(0.02mmol,9mg)分散在干燥的二氯甲烷中,加入3-苯基-5-甲氧基吲哚2(0.6mmol,133.5mg)室温搅拌2min,在冰水浴条件下加入苯磺酰氯(0.79mmol,100ul),室温反应,2h后薄层色谱检测反应完全,用硅藻土过滤,滤液减压蒸除溶剂,残余物经硅胶柱层析得到化合物3183mg。
1-苯磺酰基-3-苯基-5-甲氧基吲哚(3):v(石油醚):v(乙酸乙酯)=5:1,rf:0.47,黄色油状液体,产率84%。1hnmr(400mhz,cdcl3),δ:7.98–7.89(m,2h),7.88(d,j=4hz,1h),7.64(s,1h),7.60–7.51(m,3h),7.49–7.45(m,2h),7.45–7.40(m,2h),7.40–7.34(m,1h),7.19(d,j=2.4hz,1h),6.98(dd,j=9.0,2.4hz,1h),3.80(s,3h).13cnmr(101mhz,cdcl3)δ156.83,138.17,133.76,133.05,130.31,129.25,128.94,127.80,127.57,126.74,123.77,114.74,113.92,103.04,55.73.核磁共振氢谱(1hnmr)见图3。
1-苯磺酰基-3-苯基-5-羟基吲哚(4)的合成
取1-苯磺酰基-3-苯基-5-甲氧基吲哚(3)(0.5mmol,183mg),加入干燥的二氯甲烷2.5ml,在-78℃和氮气保护的条件下,加入三溴化硼(3ml);将反应置于-78℃的条件下反应0.5h后,将温度逐渐升至室温并用薄层色谱检测反应的情况,2.5h后薄层色谱检测反应完全。向反应液中加入48ml水稀释反应液,用饱和碳酸氢钠溶液中和反应液,用二氯甲烷萃取有机层,有机层用饱和氯化钠溶液洗并用无水硫酸钠干燥,过滤,滤液减压蒸除溶剂,残余物经硅胶柱层析纯化得到化合物4116.2mg。
1-苯磺酰基-3-苯基-5-羟基吲哚(4):黄色油状物,v(石油醚):v(乙酸乙酯)=4:1,rf:0.47,产率67%。1hnmr(400mhz,cdcl3)δ:7.92–7.87(m,2h),7.86(d,j=4hz,1h),7.63(s,1h),7.57–7.48(m,3h),7.45–7.40(m,2h),7.42–7.38(m,2h),7.37–7.28(m,1h),7.17(d,j=2.4hz,1h),6.88(dd,j=8.9,2.4hz,1h).13cnmr(101mhz,cdcl3)δ152.60,138.09,133.80,132.89,130.64,129.26,128.89,127.73,127.58,126.74,123.93,114.83,114.02,105.65.核磁共振氢谱(1hnmr)见图5。
1-苯磺酰基-3-苯基吲哚-4,5-二酮(5)的合成
取1-苯磺酰基-3-苯基-5-羟基吲哚(0.28mmol,97.8mg)溶于1.4ml乙酸乙酯中,然后加入2-碘酰基苯甲酸(0.34mmol,194mg),室温反应过夜。薄层色谱检测反应完后用硅藻土过滤,滤液减压蒸除溶剂,经硅胶柱层析的到化合物577mg。
1-苯磺酰基-3-苯基吲哚-4,5-二酮:暗红色固体,v(石油醚):v(乙酸乙酯)=2:1,rf:0.42,产率76%,m.p.135℃~146℃。1hnmr(400mhz,cdcl3)δ:7.99-7.92(m,3h),7.74(tt,j=7.5hz,1h),7.66-7.60(m,2h),7.61–7.56(m,2h),7.44(s,1h),7.41–7.34(m,3h),6.20(d,j=10.4hz,1h).13cnmr(101mhz,cdcl3)δ181.61,173.92,137.60,136.90,135.40,131.02,130.45,130.20,128.67,128.47,128.32,127.18,125.92,123.46.核磁共振氢谱(1hnmr)见图5。
- 还没有人留言评论。精彩留言会获得点赞!