一种单取代或二取代的呋喃衍生物的合成方法
1.本发明涉及医药合成领域,更具体地,涉及一种单取代或二取代的呋喃衍生物的合成方法。
背景技术:
2.呋喃衍生物的合成一直是有机合成的关注点,这是因为有许多重要药物、香料以及具有生理活性的天然产物是以呋喃为核心结构的。传统合成呋喃的经典方法是paal
‑
knorr反应,即通过1,4
‑
二羰基化合物脱水缩合得到,但该方法所用原料1,4
‑
二酮类化合物难以获得,限制了其应用。
3.分子结构较为复杂的呋喃衍生物往往依靠从天然产物中提取,这种方法成本高,收率低,对自然资源依赖性强。呋喃衍生物还可以利用呋喃环具芳香环性质,通过对其进行卤化、硝化、磺化、酰化等亲电取代反应,获得不同的取代产物。这些方法合成呋喃衍生物受呋喃环电子效应的影响,取代基团修饰的数量及位置有限。例如,很难通过亲电取代反应在呋喃环上引入芳基。
4.围绕上述技术的局限性,中国专利“一种呋喃衍生物的合成方法”(申请号:201711042943.0,公开日:2018.03.13)由芳香基乙酮化合物和二甲基亚砜在碘类催化剂催化作用下通过过硫酸盐氧化一步反应生成了呋喃衍生物。通过在芳基乙酮的芳环上引入不同取代基,可以衍生出多种3
‑
甲硫基呋喃衍生物。该方法采用常规的芳香基乙酮化合物和二甲基亚砜作为原料,相对现有的1,4
‑
二酮类化合物原料具有成本低的优点,然而,这种方法只能合成三取代的呋喃衍生物,且两个芳基必须完全相同。
5.因此,开发单取代或二取代呋喃衍生物的合成方法,能够拓展呋喃衍生物的类型,这对呋喃类药物中间体的合成具有明显的优势。
技术实现要素:
6.本发明旨在克服上述现有技术的至少一种缺陷(不足),提供一种单取代或二取代呋喃衍生物的合成方法,能够合成多种不同取代基的呋喃衍生物,具有高度灵活性。
7.本发明采取的技术方案是,一种单取代或二取代呋喃衍生物的合成方法,在酸性溶剂中,以5
‑
羟甲基
‑
δ2‑
异噁唑啉衍生物为原料,在金属还原剂作用下一步合成单取代或二取代呋喃衍生物。
8.本发明提供了5
‑
羟甲基取代的δ2‑
异噁唑啉在金属还原剂-酸溶剂体系中“一锅法”转变成呋喃衍生物的新方法,并将其扩展到多种2
‑
单取代或2,4
‑
二取代呋喃衍生物的合成。该反应经过了还原开环-水解-脱水缩合几个过程,具有收率高,简便高效的特点。
9.进一步地,包括如下步骤:
10.a.向盛有5
‑
羟甲基
‑
δ2‑
异噁唑啉衍生物的酸性溶剂中,加入金属还原剂,升温搅拌反应;
11.b.待反应完成后,冷却至室温,用水稀释,加入乙酸乙酯萃取产物,分出有机层,水
层用乙酸乙酯萃取;
12.c.合并有机层并依次用水、饱和碳酸钠溶液洗涤,干燥,过滤,蒸发溶剂,产物纯化。
13.优选地,所述步骤a中,升温至80~100℃,搅拌2~3小时;
14.或所述步骤b中,水层用乙酸乙酯萃取2~3次;
15.或所述步骤c中,具体步骤为:合并有机层并依次用水、饱和碳酸钠溶液各洗2~3次,加入硫酸镁干燥,过滤,滤液在旋转蒸发仪上减压蒸发掉溶剂,用硅胶柱层析纯化得单取代呋喃衍生物或二取代呋喃衍生物。
16.进一步地,所述酸性溶剂为醋酸溶液或稀盐酸溶液中的一种或两种。醋酸与稀盐酸可作为还原时的氢源,同时又能促使分子内缩水。
17.优选地,所述酸性溶剂为醋酸溶液。醋酸相对于稀盐酸,能够使得单取代或二取代的呋喃衍生物的产率更高。
18.进一步地,所述醋酸溶液中,醋酸与水的体积比为4:1~1:1。
19.优选地,醋酸与水的体积比3:1。
20.进一步地,所述金属还原剂为锌粉或铁粉中的一种或两种。
21.优选地,所述金属还原剂为锌粉。因铁粉易形成难处理的酸性铁泥。
22.进一步地,所述金属还原剂与5
‑
羟甲基
‑
δ2‑
异噁唑啉衍生物的摩尔比为4~10:1。
23.优选地,所述金属还原剂与5
‑
羟甲基
‑
δ2‑
异噁唑啉衍生物的摩尔比为5:1。
24.进一步地,所述5
‑
羟甲基
‑
δ2‑
异噁唑啉衍生物的结构式如下:
[0025][0026]
其中,r1为芳基、烯基、杂芳基中的一种,r2为h、烷基中的一种。
[0027]
本发明所述的单取代或二取代的呋喃衍生物的合成方法,其反应方程式如下:
[0028][0029]
本发明的反应机理如下所示:
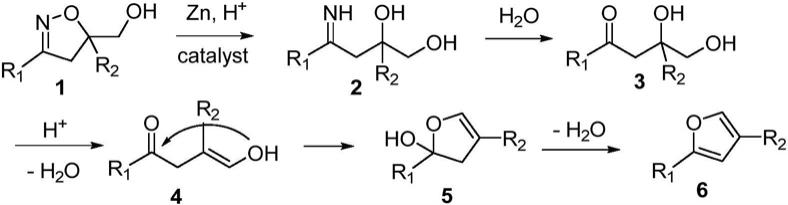
[0030][0031]
首先,δ2‑
异噁唑啉1被锌粉还原而发生n
‑
o键断裂生成亚胺中间体2,后者不稳定立即水解成β,γ
‑
二羟基酮3。在酸的作用下,β,γ
‑
二羟基酮3可以脱水得到中间体γ
‑
羰基醛(已由1hnmr检测到),它的烯醇式4进一步发生分子内的亲核加成生成关环产物5。最后,关环产物5在酸催化下再脱去一分子的水得到取代的呋喃衍生物6。在此过程中,酸的使用非常重要,它既作为机理中第一阶段金属还原时的氢源,又能够促使第二阶段分子内缩合
反应时脱水。
[0032]
与现有技术相比,本发明的有益效果为:
[0033]
1)本发明能够通过采用具有不同取代基的5
‑
羟甲基
‑
δ2‑
异噁唑啉衍生物,合成多种不同取代基的呋喃衍生物,具有高度灵活性。
[0034]
2)本发明的技术方案步骤简单、反应条件温和,通过一锅法可以实现呋喃衍生物的合成,且反应收率高,有利于大规模生产。
具体实施方式
[0035]
下面结合实施例对本发明作进一步的说明,很显然,实施例并不构成对本发明要求保护范围的限制。
[0036]
实施例1
[0037]
的合成
[0038]
向盛有5
‑
羟甲基
‑3‑
对甲苯基
‑
δ2‑
异噁唑啉(0.4mmol)的醋酸
‑
水(3∶1,8ml)溶液中加入锌粉(2.0mmol)。加毕,将反应温度升至100℃搅拌3小时。反应液冷却到室温,用水稀释。加入乙酸乙酯萃取产物。分出有机层,水层用乙酸乙酯萃取2次。合并有机层并依次用水、饱和碳酸钠溶液各洗3次,加入硫酸镁干燥,过滤,滤液在旋转蒸发仪上减压蒸发掉溶剂。残余物用硅胶柱层析(石油醚淋洗)纯化得产物2
‑
对甲苯基呋喃,产率93%。核磁数据如下:1h nmr(300mhz,cdcl3)δ7.60(2h,d,j=8.2hz),7.47(1h,s),7.22(2h,d,j=8.2hz),6.62(1h,d,j=3.3hz),6.48(1h,dd,j=3.3,1.8hz),2.39(3h,s);
13
c nmr(75mhz,cdcl3)δ154.2,141.6,137.1,129.3,128.2,123.7,111.5,104.2,21.2.
[0039]
实施例2
[0040]
的合成
[0041]
向盛有5
‑
羟甲基
‑3‑
对甲氧苯基
‑
δ2‑
异噁唑啉(0.4mmol)的醋酸
‑
水(3∶1,8ml)溶液中加入铁粉(2.0mmol)。加毕,将反应温度升至100℃搅拌2小时。反应液冷却到室温,用水稀释。加入乙酸乙酯萃取产物。分出有机层,水层用乙酸乙酯萃取2次。合并有机层并依次用水、饱和碳酸钠溶液各洗3次,加入硫酸镁干燥,过滤,滤液在旋转蒸发仪上减压蒸发掉溶剂。残余物用硅胶柱层析(石油醚淋洗)纯化得产物2
‑
对甲氧苯基呋喃,产率85%。核磁数据如下:1h nmr(300mhz,cdcl3)δ7.6l(2h,d,j=6.8hz),7.43(1h,s),6.92(2h,d,j=6.8hz),6.52(1h,dd,j=3.3,0.7hz),6.45(1h,dd,j=3.3,1.8hz),3.84(3h,s);
13
c nmr(75mhz,cdcl3)δ158.9,154.0,141.4,125.2,124.0,114.1,111.5,103.3,55.3.
[0042]
实施例3
[0043]
的合成
[0044]
向盛有3
‑
对氟苯基
‑5‑
羟甲基
‑
δ2‑
异噁唑啉(0.4mmol)的稀盐酸(0.2m,8ml)溶液中加入锌粉(2.0mmol)。加毕,将反应温度升至100℃搅拌3小时。反应液冷却到室温,用水稀释。加入乙酸乙酯萃取产物。分出有机层,水层用乙酸乙酯萃取2次。合并有机层并依次用水、饱和碳酸钠溶液各洗3次,加入硫酸镁干燥,过滤,滤液在旋转蒸发仪上减压蒸发掉溶
剂。残余物用硅胶柱层析(石油醚淋洗)纯化得产物2
‑
氟苯基呋喃,产率65%。核磁数据如下:1h nmr(300mhz,cdcl3)δ7.62
‑
7.67(2h,m),7.46(1h,dd,j=1.7,0.6hz),7.08(2h,t,j=8.8hz),6.59(1h,d,j=3.3hz),6.47(1h,dd,j=3.3,1.8hz);
13
c nmr(75mhz,cdcl3)δ163.7,160.5,153.2,142.0,127.3,125.5,115.7,111.6,104.6.
[0045]
实施例4
[0046]
的合成
[0047]
向盛有3
‑
(5
‑
氯呋喃
‑2‑
基)
‑5‑
羟甲基
‑
δ2‑
异噁唑啉(0.4mmol)的醋酸
‑
水(4∶1,8ml)溶液中加入锌粉(2.0mmol)。加毕,将反应温度升至100℃搅拌3小时。反应液冷却到室温,用水稀释。加入乙酸乙酯萃取产物。分出有机层,水层用乙酸乙酯萃取2次。合并有机层并依次用水、饱和碳酸钠溶液各洗3次,加入硫酸镁干燥,过滤,滤液在旋转蒸发仪上减压蒸发掉溶剂。残余物用硅胶柱层析(石油醚淋洗)纯化得产物2
‑
氯
‑5‑
(呋喃
‑2‑
基)呋喃,产率84%。核磁数据如下:1h nmr(300mhz,cdcl3)δ7.40(1h,dd,j=1.7,0.7hz),6.55(1h,d,j=3.3hz),6.51(1h,d,j=3.4hz),6.45(1h,dd,j=3.4,1.8hz),6.23(1h,d,j=3.4hz);
13
c nmr(75mhz,cdcl3)δ146.0,145.4,141.9,135.7,111.4,108.0,106.8,105.5.
[0048]
实施例5
[0049]
的合成
[0050]
向盛有5
‑
羟甲基
‑3‑
(2
‑
苯乙烯基)
‑
δ2‑
异噁唑啉(0.4mmol)的醋酸
‑
水(2∶1,8ml)溶液中加入锌粉(2.0mmol)。加毕,将反应温度升至100℃搅拌3小时。反应液冷却到室温,用水稀释。加入乙酸乙酯萃取产物。分出有机层,水层用乙酸乙酯萃取2次。合并有机层并依次用水、饱和碳酸钠溶液各洗3次,加入硫酸镁干燥,过滤,滤液在旋转蒸发仪上减压蒸发掉溶剂。残余物用硅胶柱层析(石油醚淋洗)纯化得产物2
‑
(2
‑
苯乙烯基)呋喃,产率90%。核磁数据如下:1h nmr(300mhz,cdcl3)δ7.48
‑
7.51(2h,m),7.43(1h,d,j=1.5hz),7.36
‑
7.39(2h,m),7.26
‑
7.29(1h,m),7.05(1h,d,j=16.3hz),6.92(1h,d,j=16.3hz),6.45(1h,dd,j=3.3,1.8hz),6.38(1h,d,j=3.3hz);
13
c nmr(75mhz,cdcl3)δ153.2,142.1,136.9,128.7,127.5,127.1,126.3,116.5,111.6,108.6.
[0051]
实施例6
[0052]
的合成
[0053]
向盛有3
‑
(邻甲氧苯基)
‑5‑
羟甲基
‑
δ2‑
异噁唑啉(0.4mmol)的醋酸
‑
水(1∶1,8ml)溶液中加入锌粉(2.0mmol)。加毕,将反应温度升至100℃搅拌3小时。反应液冷却到室温,用水稀释。加入乙酸乙酯萃取产物。分出有机层,水层用乙酸乙酯萃取2次。合并有机层并依次用水、饱和碳酸钠溶液各洗3次,加入硫酸镁干燥,过滤,滤液在旋转蒸发仪上减压蒸发掉溶剂。残余物用硅胶柱层析(石油醚淋洗)纯化得产物2
‑
(邻甲氧苯基)呋喃,产率91%。核磁数据如下:1h nmr(300mhz,cdcl3)δ7.90(1h,dd,j=7.7,1.7hz),7.50(1h,dd,j=1.7,0.7hz),7.26
‑
7.28(1h,m),7.04
‑
7.07(1h,m),6.96
‑
7.00(2h,m),6.53(1h,dd,j=3.3,1.8hz),3.96
(3h,s);
13
c nmr(75mhz,cdcl3)δ155.2,150.2,141.0,128.0,125.9,120.7,119.8,111.6,110.9,109.8,55.3.
[0054]
实施例7
[0055]
的合成
[0056]
向盛有3
‑
邻甲氧苯基
‑5‑
羟乙基
‑5‑
羟甲基
‑
δ2‑
异噁唑啉(0.4mmol)的醋酸
‑
水(3∶1,8ml)溶液中加入锌粉(2.0mmol)。加毕,将反应温度升至80℃搅拌3小时。反应液冷却到室温,用水稀释。加入乙酸乙酯萃取产物。分出有机层,水层用乙酸乙酯萃取2次。合并有机层并依次用水、饱和碳酸钠溶液各洗3次,加入硫酸镁干燥,过滤,滤液在旋转蒸发仪上减压蒸发掉溶剂。残余物用硅胶柱层析(石油醚淋洗)纯化得产物2
‑
邻甲氧苯基
‑4‑
羟乙基呋喃,产率63%。核磁数据如下:1h nmr(300mhz,cdcl3)δ7.83(1h,dd,j=7.7,1.5hz),7.33(1h,s),7.21
‑
7.26(1h,m),6.93
‑
7.04(2h,m),6.88(1h,s),3.93(3h,s),3.82(2h,t,j=6.4hz),2.72(2h,t,j=6.4hz),2.01
‑
2.07(1h,br).
[0057]
显然,本发明的上述实施例仅仅是为清楚地说明本发明技术方案所作的举例,而并非是对本发明的具体实施方式的限定。凡在本发明权利要求书的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1