一种盐酸托莫西汀的制备方法与流程
1.本发明涉及药物合成技术领域,具体涉及一种盐酸托莫西汀的制备方法。
背景技术:
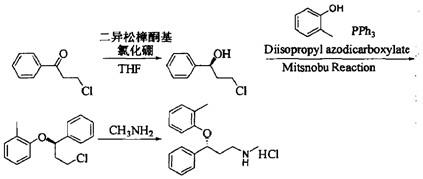
2.盐酸阿托莫西汀,化学名为:(r)
‑
n
‑
甲基
‑3‑
(2
‑
甲基苯氧基)
‑3‑
苯丙基胺盐酸盐,商品名:strattera。拜耳制药公司开发,2002年11月由美国fda(food and drugadministration)批准上市,适用于6岁及以上儿童多动症(adhd)患者的治疗;2004年由欧洲药品管理局(ema)批准上市,用于治疗儿童多动症。
3.盐酸阿托莫西汀,托莫西汀的(r)
‑
(
‑
)对映异构体,是一种选择性去甲肾上腺素再摄取抑制剂,结构式如下:盐酸阿托莫西汀的合成路线众多,如以二异松樟酮基氯化硼为催化剂,选择性还原3
‑
氯
‑1‑
苯基酮得到(s)
‑
(
‑
)
‑3‑
氯
‑1‑
苯基丙醇。而后将(s)
‑
(
‑
)
‑3‑
氯
‑1‑
苯基丙醇经mitsunobu反应得醚,再经甲胺化反应,成盐后既得产物。
4.本方法反应步骤少,立体选择性好,但选择性催化剂的价格昂贵。
5.如苯丙醇经氯代、溴化、醚化,后经甲胺化、拆分成盐制备。
6.本方法利用扁桃酸进行拆分,然后酸化成盐酸盐,最终生成r
‑
(
‑
)
‑
托莫西汀盐酸盐。该方法虽然反应条件温和,但是反应步骤多,不利于工业化生产。
技术实现要素:
7.本发明提供一种盐酸托莫西汀的制备方法,该合成路线反应步骤少,反应条件温和,易于工业化生产。
8.为达到上述技术目的,本发明的技术方案是这样的实现的:一种盐酸托莫西汀的制备方法,包括下述步骤:(1)化合物i经选择性还原生成化合物ii;(2)化合物ii与邻氟甲苯发生取代反应生成化合物iii;(3)化合物iii经甲胺化反应生成化合物iv;
(4)化合物iv在有机溶剂中与氯化氢气体成盐得到盐酸托莫西汀。
9.其中,所述步骤(1)的具体为:取nabh4加入圆底烧瓶中,充氮气,加入二甲氧基乙烷和α
‑
蒎烯,搅拌混合,冷却至
‑
10℃,滴入bcl3,室温下陈化2小时,得化合物i;室温下向三口瓶中加入异丙醚,搅拌均匀,滴加化合物i,20
‑
30℃反应两小时,反应液用无水乙醇萃取,用饱和食盐水洗涤ph值至8.0,晾干,得化合物ii。
10.其中,所述α
‑
蒎烯的光学纯度≥70%。
11.其中,所述nabh4与α
‑
蒎烯的摩尔比为1:3
‑
5,所述nabh4与bcl3的摩尔比为1:1
‑
1.2,所述nabh4与化合物i的摩尔比为:1:1
‑
1.25。
12.其中,所述步骤(2)具体为:向三口瓶中加入二甲基亚砜,然后加入化合物ii搅拌溶解,升温至80
‑
90℃,再滴加邻氟甲苯,tlc监测反应完全,冷却至室温,加入乙酸乙酯和水,充分搅拌分出有机相,用水洗涤,抽滤,收集滤液,滤液中加入草酸,抽滤收集滤饼,将滤饼40
‑
50℃减压干燥,得化合物iii。
13.其中,所述化合物ii与邻氟甲苯的摩尔比为1:1
‑
1.3。
14.其中,所述步骤(3)是将化合物iii至甲胺的乙醇溶液中回流3
‑
4小时,得化合物iv,所述化合物iii与所述甲胺的摩尔比为1:1.1
‑
1.3。
15.其中,所述步骤(4)具体为:向三口瓶中加入化合物iv,加入丙酮,搅拌溶解,控制体系温度至60
‑
70℃,通入氯化氢气体调节体系ph值1
‑
2,搅拌析晶,抽滤,将滤饼置50℃鼓风干燥,得化合物v。
16.本发明的有益效果:本发明以3
‑
氯
‑1‑
苯基丙酮为原料,经过还原反应、取代反应、甲胺化反应和成盐反应制得(r)
‑
n
‑
甲基
‑3‑
(2
‑
甲基苯氧基)
‑3‑
苯丙基胺盐酸盐,本合成路线反应步骤少,反应条件温和,操作简单,以3
‑
氯
‑1‑
苯基丙酮为原料,原料价廉易得,生产成本低。
具体实施方式
17.下面通过本发明具体实施例,对本发明的技术方案进行清楚、完整地描述,显然,
所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
18.实施例1本实施例提供一种盐酸托莫西汀的制备方法,包括下述步骤:(1)取nabh4(50g)加入圆底烧瓶中,充氮气,加入二甲氧基乙烷2000ml和α
‑
蒎烯(720.2g),搅拌混合,冷却至
‑
10℃,滴入bcl3(170.4g),室温下陈化2小时,得化合物i(256.3g);室温下向三口瓶中加入异丙醚,搅拌均匀,滴加化合物i,25℃反应两小时,反应液用无水乙醇萃取,用饱和食盐水洗涤ph值至8.0,晾干,得化合物ii(232.5g),收率为90.2%。其中,所述α
‑
蒎烯的光学纯度为78%。所述nabh4与α
‑
蒎烯的摩尔比为1:4,所述nabh4与bcl3的摩尔比为1:1.1,所述nabh4与化合物i的摩尔比为:1:1.15。(2)向三口瓶中加入二甲基亚砜1000ml,然后加入化合物ii(232.2g )搅拌溶解,升温至85℃,再滴加邻氟甲苯(180.9g),tlc监测反应完全,冷却至室温,加入乙酸乙酯500ml和水300ml,充分搅拌分出有机相,用水洗涤,抽滤,收集滤液,滤液中加入草酸,抽滤收集滤饼,将滤饼45℃减压干燥,得化合物iii(306.0g ),纯度为,收率为 80.2%。其中,所述化合物ii与邻氟甲苯的摩尔比为1:1.2。
19.(3)将化合物iii(305.0g)至甲胺的乙醇溶液中(含甲胺 40.8g)回流3.5小时,得化合物iv(256.5),收率为85.6%。所述化合物iii与所述甲胺的摩尔比为1:1.2。
20.(4)向三口瓶中加入化合物iv(251.1g),加入丙酮1000ml,搅拌溶解,控制体系温度至65℃,通入氯化氢气体调节体系ph值1.5,搅拌析晶,抽滤,将滤饼置50℃鼓风干燥,得化合物v(211.2g),纯度为99.2%,收率为78.3%。
21.实施例2本实施例提供一种盐酸托莫西汀的制备方法,包括下述步骤:(1)取nabh4(50g)加入圆底烧瓶中,充氮气,加入二甲氧基乙烷2000ml和α
‑
蒎烯(540.2g),搅拌混合,冷却至
‑
10℃,滴入bcl3(154.9g),室温下陈化2小时,得化合物i(222.9g);室温下向三口瓶中加入异丙醚,搅拌均匀,滴加化合物i,20
‑
30℃反应两小时,反应液用无水乙醇萃取,用饱和食盐水洗涤ph值至8.0,晾干,得化合物ii(185.6),收率为91.8%。其中,所述α
‑
蒎烯的光学纯度≥70%。所述nabh4与α
‑
蒎烯的摩尔比为1:3,所述nabh4与bcl3的摩尔比为1:1,所述nabh4与化合物i的摩尔比为:1:1。(2)向三口瓶中加入二甲基亚砜1000ml,然后加入化合物ii(184.9g)搅拌溶解,升温至80℃,再滴加邻氟甲苯(120.1g),tlc监测反应完全,冷却至室温,加入乙酸乙酯500ml和水300ml,充分搅拌分出有机相,用水洗涤,抽滤,收集滤液,滤液中加入草酸,抽滤收集滤饼,将滤饼50℃减压干燥,得化合物iii(241.9g ),纯度为,收率为79.6%。其中,所述化合物ii与邻氟甲苯的摩尔比为1:1。
22.(3)将化合物iii(240.0g)至甲胺的乙醇溶液中(含甲胺 29.5g)回流3小时,得化合物iv(19.4.0g),收率为82.3%。所述化合物iii与所述甲胺的摩尔比为1:1.1。
23.(4)向三口瓶中加入化合物iv(192.2g),加入丙酮1000ml,搅拌溶解,控制体系温度至60℃,通入氯化氢气体调节体系ph值2,搅拌析晶,抽滤,将滤饼置50℃鼓风干燥,得化合物v(162.1g ),纯度为99.3%,收率为78.6%。
24.实施例3本实施例提供一种盐酸托莫西汀的制备方法,包括下述步骤:(1)取nabh4(50g)加入圆底烧瓶中,充氮气,加入二甲氧基乙烷2000ml和α
‑
蒎烯(900.3g),搅拌混合,冷却至
‑
10℃,滴入bcl3(185.9g),室温下陈化2小时,得化合物i(278.6g);室温下向三口瓶中加入异丙醚,搅拌均匀,滴加化合物i,20
‑
30℃反应两小时,反应液用无水乙醇萃取,用饱和食盐水洗涤ph值至8.0,晾干,得化合物ii(207.9g),纯度为,收率为89.6%。其中,所述α
‑
蒎烯的光学纯度≥70%。所述nabh4与α
‑
蒎烯的摩尔比为1:5,所述nabh4与bcl3的摩尔比为1: 1.2,所述nabh4与化合物i的摩尔比为:1: 1.25。(2)向三口瓶中加入二甲基亚砜1000ml,然后加入化合物ii(206.7g )搅拌溶解,升温至90℃,再滴加邻氟甲苯(174.5g),tlc监测反应完全,冷却至室温,加入乙酸乙酯500ml和水300ml,充分搅拌分出有机相,用水洗涤,抽滤,收集滤液,滤液中加入草酸,抽滤收集滤饼,将滤饼40℃减压干燥,得化合物iii(275.8g ),纯度为,收率为81.2%。其中,所述化合物ii与邻氟甲苯的摩尔比为1:1.3。
25.(3)将化合物iii(275.0g)至甲胺的乙醇溶液中(含甲胺 39.9g )回流4小时,得化合物iv(228.6g),收率为84.6%。所述化合物iii与所述甲胺的摩尔比为1: 1.3。
26.(4)向三口瓶中加入化合物iv(225.6g,),加入丙酮1000ml,搅拌溶解,控制体系温度至70℃,通入氯化氢气体调节体系ph值1,搅拌析晶,抽滤,将滤饼置50℃鼓风干燥,得化合物v(191.5g, ),纯度为99.4%,收率为79.2%。
27.实施例4本实施例提供一种盐酸托莫西汀的制备方法,包括下述步骤:(1)取nabh4(50g)加入圆底烧瓶中,充氮气,加入二甲氧基乙烷300ml和α
‑
蒎烯(630.2g),搅拌混合,冷却至
‑
10℃,滴入bcl3(173.5g),室温下陈化2小时,得化合物i(278.6g);室温下向三口瓶中加入异丙醚,搅拌均匀,滴加化化合物i,20
‑
30℃反应两小时,反应液用无水乙醇萃取,用饱和食盐水洗涤ph值至8.0,晾干,得化合物ii(207.9g),收率为89.9%。其中,所述α
‑
蒎烯的光学纯度≥70%。所述nabh4与α
‑
蒎烯的摩尔比为1:3.5,所述nabh4与bcl3的摩尔比为1:1.12,所述nabh4与化合物i的摩尔比为:1:1.2。(2)向三口瓶中加入二甲基亚砜300ml,然后加入化合物ii(179.0g)搅拌溶解,升温至85℃,再滴加邻氟甲苯(127.8g),tlc监测反应完全,冷却至室温,加入乙酸乙酯500ml和水300ml,充分搅拌分出有机相,用水洗涤,抽滤,收集滤液,滤液中加入草酸,抽滤收集滤饼,将滤饼50℃减压干燥,得化合物iii(238.0g ),纯度为,收率为80.9%。其中,所述化合物ii与邻氟甲苯的摩尔比为1:1.1。
28.(3)将化合物iii(235.0g)至甲胺的乙醇溶液中(含甲胺 30.2g)回流3小时,得化合物iv(193.7g),收率为83.9%。所述化合物iii与所述甲胺的摩尔比为1:1.1
‑
1.3。
29.(4)向三口瓶中加入化合物iv(190.5g),加入丙酮1000ml,搅拌溶解,控制体系温度至65℃,通入氯化氢气体调节体系ph值2,搅拌析晶,抽滤,将滤饼置50℃鼓风干燥,得化合物v(159.0g, ),纯度为99.2%,收率为77.9%。
30.以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1