基于细菌16SrRNA基因序列的细菌“种”水平检测和分析方法与流程
基于细菌16s rrna基因序列的细菌“种”水平检测和分析方法
技术领域
1.本发明公开了一个检测分析人粪便标本的细菌16s rrna基因v3
‑
v4区序列,可从“种“水平检测和注释肠道菌群组成多样性和构成比分析方法,特别是能够检测在数量和比例上占优势的尚未分离和研究的未知细菌,属于微生物生态学、微生物分类学和微生物组学技术领域。
背景技术:
2.微生物组研究开展以来,很多研究提示人的生长发育、营养代谢、疾病状态、免疫反应等和肠道菌群相关,如结直肠癌、肥胖、糖尿病等。可是,人肠道菌群究竟包含多少个“种”(species)?各个“种”的丰度如何?迄今尚无明确答案。过去研究肠道菌群的多样性,主要靠分离培养技术体系。由于所使用的培养基和培养条件有选择性,如培养温度、氧含量、氨基酸和碳水化合物成分、盐浓度等,人们只能获得能够在这些培养基和培养条件生长起来的细菌。忽略了大量的不能够在这些培养基和培养条件生长起来的,暂时还没有能够分离、培养和鉴定的细菌,产生了很多错误信息。
3.据估计地球上大约有10
12
种原核生物,其中主要是细菌。细菌分类学层级包括界、门、纲、目、科、属和种。“种”是细菌的最低分类学单位。医学最常涉及的细菌分类学单位是“属”和“种”。一个细菌的“属”,可包括几个(如埃希氏菌属,包括6个“种”)或几百个“种”的细菌(如链球菌属,包括200多个“种”)。同一个属的不同“种”的细菌,生物学和医学意义差别很大,有的是益生菌(如嗜热链球菌[streptococcus thermophilus]),有的是致病菌(如猪链球菌[streptococcus suis])。因此,对肠道菌群的分类学多样性和构成比的信息,仅仅局限在“属”的水平,是远远不够的,容易产生误导。只有实现“种”水平的分析,才能较好揭示肠道菌群多样性和构成比变化与健康、疾病等的相关性,才有比较清晰的医学参考价值。
[0004]
所有细菌都有16s rrna,它是核糖体小亚基上的一种核糖体rna,参与蛋白质合成等过程,是细菌演化中的分子钟。16s rrna在细菌基因组中对应的基因序列,即为细菌16s rrna基因,长度约为1500碱基,由9个可变区(variable region,v1
‑
v9)和保守区序列交替组成。16s rrna基因保守区序高度保守,而可变区序列则因种属而异,且变异程度与细菌的系统发生位置(分类学上的种、属、科等)密切相关。因此,使用16s rrna基因序列分析,可将所有细菌进行鉴定分类。如使用16s rrna基因全长序列,在大多数情况下可将待测细菌鉴定到“种”的水平。
[0005]
使用部分16s rrna基因序列,如v3
‑
v4区段序列,可将研究较多的、公共数据库中具有16s rrna基因序列已知细菌分类到“种”;由于缺乏参比序列,只能将大部分未知细菌分类到“属”、“科”等高阶分类学单元。少数情况下,因为一些细菌“种”的全长16s rrna基因非常相似,仅仅依靠16s rrna基因,无法准确鉴定到“种”。通常把这几个不能使用全长16s rrna基因区分的“种”,划为一个群(group)。
[0006]
16s rrna基因序列分析已经成为细菌检测鉴定和菌群多样性分析的重要方法。随
着测序技术的发展和成本降低,基于二代测序平台的高通量测序,可不依赖于细菌培养,获得海量的细菌16s rrna基因序列,为研究菌群多样性提供了有力的工具。其中肠道菌群多样性分析常用的方法,是对粪便标本进行基于illumiina测序平台对16s rrna基因v3
‑
v4区(400碱基左右)开展高通量测序,获得海量序列。单个样本将获得十万及以上条16s rrna基因序列,经序列比对分析和注释等环节,最终完成样本中肠道(粪便)菌群的细菌分类学分析和鉴定。获得肠道菌群多样性(含有多少“种”或“属”的细菌)和构成比(每个“种”或“属”的细菌,占所有序列数的百分比)的数据。由于大量的肠道菌群是未知细菌,尚未分离鉴定,缺乏相应的全长16s rrna基因序列可供比对。因此,现有肠道菌群分析技术只能将这些数量占优势的未知细菌,鉴定到“属”或“属”以上的水平,无法精确鉴定到“种”。
[0007]
现有技术的不足:使用二代测序技术扩增的16s rrna基因v3
‑
v4区等序列,仅有400碱基左右,可将大部分序列鉴定到“属”或“属”以上的分类学水平,获得“属”或“属”水平以上的肠道菌群多样性和构成比数据。这些“属”或“属”以上水平的分析数据,无法准确揭示肠道菌群的变化和健康疾病的关系,限制了肠道菌群分析的应用和推广。本发明的目的就是提供一种在“种”(species)水平上检测、鉴定、分析人体菌群的方法。
技术实现要素:
[0008]
基于上述目的,本发明首先提供了一种非诊断目的的基于细菌全长或接近全长16s rrna基因序列在“种”(species)水平上鉴定人体菌群的方法,该处所述的16s rrna基因是指全长或者接近全长的16s rrna基因序列,长度在1450
‑
1500碱基之间,所述方法包括以下步骤:
[0009]
(1)构建基于细菌操作系统发生学单元(opu,operational phylogenetic unit)为基本注释单位的人体肠道菌群16s rrna基因参比序列库。opu包括所有已知细菌,和由本发明发现的人肠道众多尚未发现的未知细菌。所述的参比序列库包括已获得“种”水平命名的所有已知细菌,和未知细菌。对于已获得现有技术命名的opu(已知细菌),采用命名的名称注释;对于未获得现有技术命名的opu,采用所述opu及其编码,及其高一级分类学单元,作为该细菌的唯一命名。本发明据此构建了人肠道菌群16s rrna基因全长参比序列库。该数据库包括所有已经被命名的细菌,和本发明发现的肠道未知细菌。所有已经被命名的已知细菌的参考菌株的16s rrna基因序列来自于已被公开的参比序列库,包括但不限于:原核生物标准命名名录、美国国立生物技术信息中心和细菌16s rrna基因序列在线质控和比对数据库收录和公开的16srrna基因序列库;
[0010]
(2)构建细菌16s rrna基因v3
‑
v4区参比序列库,对上述人体肠道菌群全长16s rrna基因参比序列库的v3
‑
v4区序列,使用计算机进行虚拟剪切,获得v3
‑
v4区序列。虚拟剪切采用16s rrna基因v3
‑
v4区通用扩增引物341f(seq id no.1)和806r(seq id no.2)的结合位点。将序列完全相同的条目进行合并后,形成肠道菌群16s rrna基因v3
‑
v4区参比序列工作库。可用于检测鉴定所有已知细菌(18000余个种)和本发明发现的健康人肠道菌群的未知菌(774个opu);
[0011]
(3)对待检测标本的16s rrna基因进行序列测定,在本发明中的一个具体的技术方案中,针对16s rrna基因v3
‑
v4区进行序列测定;
[0012]
(4)将步骤(3)获得的标本16s rrna基因序列作为查询序列,与步骤(2)肠道菌群
16s rrna基因v3
‑
v4区参比序列工作库,进行查询比对及菌种鉴定。将与参比序列工作库中带有分类学信息的特定序列完全一致(100%)的查询序列,鉴定为参比序列工作库中特定序列注释名称。在本发明中的一个具体的技术方案中,将从待测标本获得的16s rrna基因v3
‑
v4区序列,与16s rrna基因v3
‑
v4区参比序列库进行比对,对于与参比序列工作库中已知细菌的“种”16s rrna基因v3
‑
v4区参比序列一致性为100%的序列,注释为已知细菌的分类学“种”名;对于与参比序列库中的未知细菌16s rrna基因v3
‑
v4区参比序列一致性为100%的序列,注释为未知细菌,赋予唯一的opu编号。未知细菌包括疑似新种和高分阶单元。所述高分阶单元是指,仅仅依靠16s rrna基因序列难以准确鉴定,用上一级分类学单元,和opu编码表示。
[0013]
在一个优选的实施方案中,所述方法还包括对步骤(3)所鉴定菌种在待测标本中菌群种类、比例、和/或丰度分析的步骤。在具体应用中,可根据需要提供,包括但不限于,待分析标本包含opu的数目,已知菌数目、种类、丰度,未知菌的种类、数目和丰度;以及各个“种”或opu占肠道菌群总数的百分比;以及益生菌的种类和丰度,致病菌、推荐致病菌的种类和丰度,优势opu的数目和丰度等。
[0014]
在另一个优选的实施方案中,所述方法中16s rrna基因序列为v3
‑
v4区序列。本发明方法可以用于基于16s rrna基因v3
‑
v4区的菌群鉴定分析,但并不限于v3
‑
v4区,也可以用于基于16s rrna基因其它区域的菌群鉴定分析。
[0015]
在一个优选的实施方案中,所述人体菌群来源于消化道、皮肤、口腔、鼻咽部、眼部、阴道、泌尿道、耳部的菌群。
[0016]
在另一个优选的实施方案中,所述方法步骤(2)序列测定为高通量测序,本发明的一个具体实施方案是基于illumina二代测序平台对待检肠道或粪便样本进行16s rrna基因v3
‑
v4区深度测序获得序列的。
[0017]
其次,本发明提供了一种构建上述基于细菌全长或接近全长16s rrna基因序列分析的、在“种”(species)水平上检测鉴定人肠道菌群的方法中步骤(1)所述的基于细菌操作系统发生学单元为单位的人肠道菌群16s rrna基因v3
‑
v4区序列参比序列库的方法,所述方法包括:
[0018]
(1)测序和质控:获得来自人标本中的细菌16s rrna基因序列,经过质控删除低质量(如单碱基质量值低于10的序列;无法识别到双端引物的序列;嵌合体(chimeras)序列等)序列;本发明中,发明人应用三代测序技术pacbio测序平台对来源于120个健康人群的粪便标本获得了人肠道菌群16srrna基因全长或近似全长(1450
‑
1500碱基)的序列,获得850,935条16s rrna基因序列。
[0019]
使用pacbio smrt link(version 6.0.0)进行质控分析。根据rsii_384_barcodes进行样品拆分,最小条码得分(minimum barcode score)设置为26。利用环化纠错(circular consensus sequencing,ccs)的方法以降低序列的错误率,设置参数为最低5个ccs循环和最低预测准确性(minimum predicted accuracy)高于99.9%。随后,使用qiime软件进行模糊的碱基、低质量的序列、引物和测序接头的过滤。去除长度在1200~1600bp之外的序列。在本发明的一个具体实施例中,发明人使用生物信息学分析软件usearch(http://www.drive5.com/usearch/)的嵌合体检测软件uchime qiime(全称:quantitative insights into microbial ecology),筛选出594,075条全长或接近全长的
16s rrna基因序列;
[0020]
(2)划分细菌分类学操作单元(operational taxonomic unit,otu):将来自步骤(1)的序列一致性达到98.7%及以上的的一组16s rrna基因序列,划分为一个otu(每个粪便标本可获得若干otu,每个otu包含若干16s rrna基因序列);
[0021]
(3)确定每个otu(细菌分类学操作单元)的代表性序列:把在步骤(2)获得的一个细菌分类学操作单元中出现频率高居前10的16s rrna基因序列,选为该组细菌分类学操作单元的代表性序列,不足10条序列者全部选为该细菌分类学操作单元的代表性序列;
[0022]
(4)构建细菌系统发生树:使用步骤(3)获得的每个otu代表性序列和已经被命名的细菌参考菌株16s rrna基因序列进行比对,将比对上的otu代表性序列,插入到所有已经被命名的所有细菌参考菌株16s rrna基因序列数据库中,参数设置为ltp50。将插入的otu代表性序列和已经被命名的细菌参考菌株的16s rrna基因序列,使用基于jukes
‑
cantor修正的邻接法(neighbor
‑
joining method)构建所有细菌系统发生树,保守度设为30%。
[0023]
在本发明的一个具体实施方案中,所述细菌系统发生树的构建步骤为:使用步骤(3)获得每个otu的代表性序列,使用sina软件(version 1.2.11),与所有已知细菌的16s rrna基因序列(ltp132数据库)进行比对。利用arb软件(version 6.0.6)内置的parsimony工具,将比对上的otu代表性序列,插入到所有已经被命名的所有细菌参考菌株16s rrna基因序列数据库(ltp 132数据库和nr silva ref 132数据库中),参数设置为ltp50。将插入的otu代表性序列和已经被命名的细菌参考菌株的16s rrna基因序列,使用基于jukes
‑
cantor修正的邻接法(neighbor
‑
joining method)构建所有细菌系统发生树,保守度设为30%。
[0024]
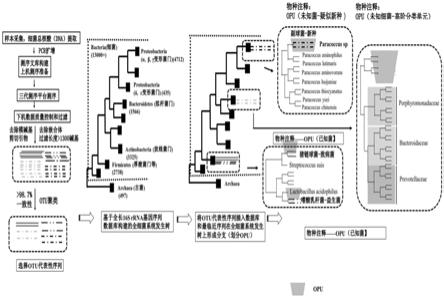
(5)发现健康人肠道未知细菌:在构建的所有细菌系统发生树上,查询otu的代表性序列会和相似度最近的16s rrna基因序列聚集,在树上形成一个分支(图1),将这个分支确定为一个opu(操作系统发生学单元)。如果otu的代表性序列与在所有细菌系统发生树上最临近的16s rrna基因序列的一致性达98.7%或以上,且已经获得命名,可使用获得命名的细菌名称注释。这类opu可确定为已知细菌(图1)。如果otu代表性序列及其在所有细菌系统发生树上最临近的16s rrna基因序列的一致性为98.7%以下,但和“属”内其他“种”的代表性序列的一致性达95%或以上,可确定为未知细菌的疑似新种(图1);如果otu代表性序列及其在所有细菌系统发生树上最临近的参考菌株的16s rrna基因序列的一致性为95%以下,且尚未获得命名,可命名为未知细菌的高分阶单元,使用编号的高一级的分类学单元和opu编号(opu number)命名(图1)。
[0025]
(6)构建基于opu(细菌操作系统发生学单元)的人肠道菌群16s rrna基因序列参比序列库:在基于已知细菌16s rrna基因构建的细菌系统发生树上,查询序列会和分类学上最临近的参考序列聚类,在所有细菌系统发生树上形成一个独立分支(树枝),命名为一个opu(图1)。查询序列和最临近的参考序列的相似度达98.7%及以上的opu,可确定为已知细菌;查询序列和最临近的参考序列的相似度低于98.7%,可确定为未知细菌。对于已获得现有技术命名的opu,为已知细菌,采用命名名称注释;未获得命名的opu,是为未知细菌,采用所述opu及其编码作为该细菌的唯一命名;
[0026]
在本发明的一个具体实施例中,通过该步骤,对本发明获得的健康人肠道细菌来源的59.4万余条全长或接近全长(1450
‑
1500碱基)的16s rrna基因序列整理获得1235个操
作系统发生学单元。这1235个opu包括461个“种”的已知细菌、774种未知细菌;
[0027]
(7)对步骤(5)获得的16s rrna基因序列参比序列库进行剪切,将序列完全相同的条目进行合并后,形成肠道菌群16s rrna基因v3
‑
v4区参比序列工作库。
[0028]
在一个优选的实施方案中,步骤(1)所述测序采用用三代测序pacbio技术平台进行,至少包括120名健康人粪便标本进行细菌16s rrna全长基因序列测定,质控中删除的低质量序列包括单碱基量值(quality)值低于10的序列、无法识别到双端引物的序列、嵌合体(chimeras)。在本发明中的一个具体的实施方案中,对120名健康人粪便标本进行细菌16s rrna全长(1450
‑
1500碱基)基因序列测定。
[0029]
在一个优选的实施方案中,步骤(4)所述已经被命名的细菌参考菌株16srrna基因序列来自于已被公开的参比序列库,所述参比序列库包括,但不限于:原核生物标准命名名录、美国国立生物技术信息中心和细菌16s rrna基因序列在线质控和比对数据库收录和公开的16s rrna基因序列库。其中,所述原核生物标准命名名录(lpsn:https://www.bacterio.net/)和美国国立生物技术信息中心(ncbi refseq database:https://www.ncbi.nlm.nih.gov/)目前公开的已知细菌参考菌株的16s rrna基因序列,合计38,000余条,包括18 000余个已经发表并认可的细菌种和亚种参考菌株的序列。所述参比序列库还吸纳细菌16s rrna基因序列在线质控和比对数据库(silva,https://www.arb
‑
silva.de/)的细菌分类学名称相同的16s rrna基因序列,兼并碱基(是指根据密码子的兼并性,用一个符号代替某两个或者更多的碱基。如兼并碱基n,可代表u/c/a/g四个碱基)比例小于2%、一致性99%以上、长度大于1000碱基以上的高质量序列,截止目前合计14.3万条。这部分序列主要来源于非参考菌株。作为已知细菌的分类学参考菌株的16s rrna基因序列的补充,提高多样性、覆盖率。本处所述的三个在线数据库均为开放性的公共数据库,并不构成对本发明数据库来源和构建方法的限制,只要能够提供细菌来源的多样性、覆盖率的数据库均可以被本发明方法所采用。本发明通过对上述3个或者以上的数据库的序列整合,构成肠道细菌16s rrna基因参比序列库,包括80余万条(包括120名健康人肠道菌群发现的、原核生物标准命名名录、美国国立生物技术信息中心和细菌16s rrna基因序列在线质控和比对数据库收录和公开的16s rrna基因序列库)16s rrna基因序列。所述80余万条16s rrna基因序列的数量并不构成对本发明数据库大小和构建方法的限制,只要能够提供细菌来源的多样性、覆盖率的数据库均可以被本发明方法所采用。
[0030]
在一个优选的实施方案中,步骤(6)所述的剪切采用16s rrna基因v3
‑
v4区计算机虚拟剪切序列。
[0031]
更为优选地,所述虚拟剪切采用的上游剪切位点的序列如seq id no.1所示(cctaygggrbgcascag),下游剪切位点的序列如seq id no.2所示(ggactacnngggtatctaat)。上述方法中步骤(6)所述的剪切采用16s rrna基因v3
‑
v4区通用扩增引物341f(seq id no.1)和806r(seq id no.2)的结合位点,进行计算机虚拟剪切,获得所有肠道菌群参比序列的v3
‑
v4区序列。将序列完全相同的条目进行合并后,形成肠道菌群16s rrna基因v3
‑
v4区参比序列工作库,包括27.3万条16s rrna基因v3
‑
v4序列,可检测鉴定所有公开的18000余个已知细菌和健康人肠道菌群的未知菌。
[0032]
现有技术中,使用16s rrna基因v3
‑
v4区高通量测序技术原理检测肠道菌群的方法,只能检测已知细菌,无法检测未知细菌。本发明通过opu的定义、发现、注释以及基于opu
的细菌系统进化树的构建,解决了上述技术难题,不仅可以对未知细菌进行检测,并使用opu来描述和注释,还可以在未知细菌的发现及致病性和治疗性应用上进行分析和预测,极大地提供了细菌鉴定、致病菌发现和益生菌筛选发明的工作效率。通过本发明提供的方法,发现人肠道菌群有774“种”未知细菌,即774个opu。特别是发现60%以上中国人粪便菌群共享116个opu,包括38种已知菌、78种未知菌(以编码的opu表示),约占菌群总数的83.42%。使用我们发现的肠道未知细菌的全长16s rrna基因序列做分类学参照,可以实现对肠道菌群未知细菌的检测,这是目前任何一种现有技术都无法实现的。
[0033]
本发明通过比较上述未知菌和已知菌的16s rrna基因序列,可将粪便标本平均95%以上16s rrna基因v3
‑
v4区高通量测序数据,鉴定为已知细菌和未知细菌(opu)。基于v3
‑
v4区序列的鉴定率,从现有技术的37.8%提高到95.6%及以上。本发明方法,可从“种”的水平,分析健康人肠道菌群失调情况;可发现已知病原菌和潜在致病菌,可分析肠道益生菌的种类和丰度,特别是肠道菌群和健康状况、疾病的关系,可用于人肠道菌群多样性、健康状态、疾病状态等的评估,包括患者肠道菌群多态性和构成比分析。
附图说明
[0034]
图1.细菌操作系统发生学单元(opu)划分技术路线图;
[0035]
图2.健康人肠道常驻菌群的116种细菌(opu)的构成比阈值;
[0036]
图3.健康人肠道常驻菌群的116种细菌(opu)的构成比阈值;
[0037]
图4.健康人肠道常驻菌群的116种细菌(opu)的构成比阈值;
[0038]
图5.健康中国人粪便菌群的多样性(种类数目)和丰度(构成比);
[0039]
图6.成人腹泻病人(f32)粪便菌群结构及丰度;
[0040]
图7.肝硬化患者(f54)粪便菌群结构及丰度;
[0041]
图8.婴儿腹泻病人(f181)粪便菌群结构及丰度。
具体实施方式
[0042]
下面结合具体实施例来进一步描述本发明。本发明的优点和特点将会随着描述而更为清楚。但这些实施例仅是范例性的,并不对本发明的权利要求所限定的保护范围构成任何限制。
[0043]
构建实施例1.肠道菌群16s rrna基因v3
‑
v4区参比序列工作库的构建
[0044]
1.构建肠道菌群16s rrna基因参比序列库
[0045]
(1)获得健康人肠道细菌来源的1235个opu的16s rrna基因序列
[0046]
对120个健康中国人肠道菌群标本,使用pacbio测序平台测序,获得850,935条16s rrna基因序列。使用pacbio smrt link(version 6.0.0)进行质控分析。利用环化纠错(circular consensus sequencing,ccs)的方法以降低序列的错误率,设置参数为最低5个ccs循环和最低预测准确性(minimum predicted accuracy)高于99.9%。随后,使用qiime软件进行模糊碱基、低质量的序列、引物和测序接头的过滤。去除长度在小于1200碱基和长于1600碱基的序列,获得594,075条全长或接近全长的16s rrna基因序列。划分为1235个opu。每个opu可包括多条频率较高的代表性16s rrna基因序列,作为参考序列,其一致性达99%及以上。
[0047]
opu是细菌操作系统发生单元的英文缩写,是分类学上最小的单系类群(monophyletic group),包括一群全长16s rrna基因序列,代表一群细菌菌株。每个opu群内菌株的16s rrna基因序列,相互之间的亲缘关系最近,属于一个单系类群。不同的opu,属于不同的单系类群。opu数量众多,包括公开发表的已知细菌和未知细菌。已知细菌可用国际细菌分类学委员会通过原核生物标准命名名录公布的名称进行注释,如肺炎链球菌。未知细菌使用本发明编号的opu进行注释,代表一个新“种”、新“属”、新“科”、新“目”、新“纲”、新“门”等。仅仅依靠全长16s rrna基因序列分析,按照目前的分类学认知,无法准确发现和定义一个新“属”及以上的分类学单元。
[0048]
opu的划分包括二个步骤:一是划分otu,二是划分opu。具体做法如下:
[0049]
1)全长16s rrna基因测序。利用三代测序平台(pacbio rs ii platform),对粪便样本中的16s rrna基因(v1
‑
v9)进行测序,获得全长或接近全长的序列(1450
‑
1500碱基)。
[0050]
2)测序数据质控。使用生物信息学分析软件usearch(http://www.drive5.com/usearch/)的嵌合体检测软件uchime qiime(全称是quantitative insights into microbial ecology),去除模糊碱基、嵌合体。此为常规方法。
[0051]
3)划分otu使用usearch软件的otu聚类和代表性序列鉴定算法划分otu。将所有一致性达到98.7%的16s rrna基因序列,划为一个otu。将每个otu中出现频率最高的前10条16s rrna基因序列,选择为这个otu的代表性序列。如果出现频率最高的16s rrna基因序列不到10条,则全部纳入。
[0052]
4)将比对上的某个otu的代表性16s rrna基因序列,鉴定为已知细菌。将查询otu的代表性16s rrna基因序列,加入所有已知细菌系统发生树(the all
‑
species living tree)数据库ltp 123,使用16s rrna序列在线查询软件sina(the new silva(web)aligner)进行序列比对。能够比对上的序列(一致性为98.7%或以上),可插入到所有已知细菌系统发生树上。基于序列对比以及系统发生树的拓扑结构和相互关系,如果能够划归为某已知细菌的16srrna基因序列,和其形成独立的分支,则可注释为某个已知细菌。如猪链球菌(steptococcus suis)。这个已知细菌,可在系统发生树上形成独立的分支,是一个有分类学名称的opu。
[0053]
5)将和所有已知细菌的参考菌株的16s rrna基因序列一致性低于98.7%的otu,鉴定为未知细菌,使用opu方法进行注释。将一致性低于98.7%的otu的代表性16s rrna基因序列,加入silva数据库的非冗余(silva reference non redundant)数据库(silva ssuref_nr_132),进行二次比对。
[0054]
将二次比发现的数据库中和查询序列一致性最接近的16s rrna基因序列,以及查询otu的代表性16s rrna基因序列,和ltp128数据库所有已知细菌参考菌株16s rrna基因序列,使用在线查询软件sina,使用邻位相接法(neighbor
‑
joining),构建所有细菌系统发生树。设定古菌为树根(root)(图1)。
[0055]
分析形成的所有细菌系统发生树的拓扑结构,定义每一个opu。每个opu都是最小的单系类群(monophyletic group)。每个opu都至少包括二类序列:otu的代表性序列,和这些代表性序列最接近的16s rrna基因序列,特别是最接近的参考菌株的16s rrna基因序列(图1)。
[0056]
6)可注释为疑似新种的opu。如果一个opu可以鉴定到某个“属”,但是和“属”内所
有“种”的参考菌株的16s rrna基因序列的一致性均低于98.7%,可注释为一个未知新种细菌。
[0057]
7)高分阶单元opu的注释。如果依据细菌系统发生树,只能够把某个opu鉴定到“科”,或者“科”以上的分类学单元,我们把它作为未知高分阶单元对待,可认为至少代表一个未知“属”。因为,无法仅仅依据全长16s rrna基因序列,正确做出“种”以上水平的分类学鉴定(图1)。
[0058]
8)opu编号。所有opu统一编号。每个opu的编号都是唯一的。
[0059]
在120名健康人粪便标本中,使用上述方法,划分了1235个opu。其中,461个opu可鉴定为已知细菌,可鉴定到“种”;774个opu(62.7%)是未知细菌。在774个未知细菌opu中,有358个可鉴定到属,注释为某个“属”的疑似新种。其余416个opu,无法准确鉴定,注释为“高水平分类单元(图1)。
[0060]
从中国120个健康人粪便标本获得的全长或接近全长的16s rrna基因序列中,54.45%属于未知细菌,尚未分离、命名、研究。提示,50%以上的肠道菌群是未知细菌。
[0061]
在健康中国人肠道菌群1235个opu中,有116个opu可以在60%以上的粪便标本检测到。其中,只有38个opu是已知细菌,78个opu(67%)是未知细菌3。图2展示了检出率为60%及以上的116种细菌的构成比及其差异范围。没有一种细菌的检出率为100%。不同健康个体肠道菌群的构成不是完全一致的,差异很大,但有相似性。我们把检出率为60%及以上的116种细菌,称之为中国人肠道常驻菌群(图2),是肠道菌群维持平衡的主要成员。其中,已知细菌用细菌认可名称表示,如prevotella copri。未知细菌用opu及编码表示,如bacteroides sp.17(opu
‑
532),表示拟杆菌属的一个疑似新种,尚未分离鉴定;如lachnospiraceae(opu
‑
001),表示lachnospiraceae(毛螺菌科)中的一个新成员,仅仅依靠16s rrna基因序列难以准确鉴定,称之为高分阶单元opu。
[0062]
(2)获得所有已知细菌参考菌株的参考16s rrna基因序列。包括原核生物(主要是细菌)标准命名名录(lpsn:https://www.bacterio.net/)和美国国立生物技术信息中心(ncbi refseq database:https://www.ncbi.nlm.nih.gov/)已知细菌参考菌株的16s rrna基因序列,合计38,000余条。每个细菌“种”,可包括多条16s rrna基因序列。
[0063]
(3)扩展上述已知细菌参考菌株的参考16s rrna基因序列库。吸纳16srrna基因序列质量核查和比对在线数据库silva(https://www.arb
‑
silva.de/)的、分类学名称完全一致的、兼并碱基比例小于2%、长度1000bp以上的、一致性大于99%的高质量序列,合计14.3万条。作为公共数据库的已知细菌参考菌株的16s rrna基因序列的补充,提高灵敏度、覆盖率和准确性。
[0064]
(4)构建肠道菌群16s rrna基因参比序列库。将本发明发现的健康人肠道细菌来源的1235个opu的16s rrna基因序列、所有原核生物标准命名名录列出的已知细菌的参考菌株的16s rrna基因序列、silva数据库的已知细菌的高质量16s rrna基因序列,进行整合,构建肠道菌群16s rrna基因参比序列库。包括85万条的高质量细菌16s rrna基因,可检测、鉴定所有公布的18,000余个细菌种和亚种。特别是能够检测鉴定774种未知细菌。有库容量大、序列长度长、分类注释信息准确的特点。同时,根据新种细菌的发现和发表情况,进行更新。实现能够检测、鉴定所有已知细菌的目标(图1)。
[0065]
2.肠道菌群16s rrna基因v3
‑
v4区参比序列工作库的构建
[0066]
将我们构建的肠道细菌16s rrna基因参比序列库中85万条序列,按照16s rrna基因v3
‑
v4区扩增引物341f(cctaygggrbgcascag)和806r(ggactacnngggtatctaat)的结合位点,进行计算机剪切,获得所有85万条16s rrna基因的v3
‑
v4区序列。即对参比序列库中每一条全长16s rrna基因,进行计算机虚拟剪切,保留v3
‑
v4区序列,组成肠道菌群16s rrna基因v3
‑
v4区参比序列工作库。在新组建的参比序列工作库中,将完全相同的序列条目合并。本实施例构建的包括27.3万条16s rrna基因v3
‑
v4序列,可检测、鉴定18,000余个细菌种和亚种。由于包括了健康人肠道未知细菌的16s rrna基因序列,能够将大多数从人粪便标本获得的细菌16s rrna基因v3
‑
v4序列,鉴定到细菌的“种“。
[0067]
本发明构建的细菌16s rrna基因v3
‑
v4序列是一个动态的数据库,可根据在线的公开数据库,以及研究者自行研究获得的数据库的增长而发生变动,但是数据库的变动不影响本发明方法的实施,而且随着数据库的增长,对基于细菌16s rrna基因序列在“种”(species)水平上鉴定人体菌群的准确性会有相应的提高,本发明的核心并不在于数据库本身的构成,而在于构建一种动态和开放的基于细菌操作系统发生学单元为单位的人体菌群16s rrna基因序列参比序列库方法。
[0068]
构建实施例2.“种”水平肠道菌群组成多样性和构成比分析方法的构建
[0069]
在实施例1构建的数据库的基础上(图1),对待检测样本进行“种”水平的肠道菌群组成多样性和构成比分析方法或系统的构建。
[0070]
具体实施方案包括4个部分:粪便标本采集和处理、16s rrna基因v3
‑
v4区高通量测序、“种”水平的分类学注释、人粪便菌群多样性和构成比结果呈现。
[0071]
1.标本的采集和处理
[0072]
用便杯采集新鲜的粪便标本,临时存放于冰袋样本箱中,随后冷链转运至实验室,进行核酸提取。提取方法采用柱纯化粪便核酸提取试剂盒(qiagen,cat.51604),取200mg粪便样本,按说明书方法进行提取。最后用200μl去离子水洗脱离心柱搜集粪便核酸,用于后续16s rrna基因扩增。
[0073]
2. 16s rrna基因v3
‑
v4区高通量测序
[0074]
粪便核酸经pcr扩增、产物纯化,使用illumina miseq平台进行16s rrna基因v3
‑
v4区进行双端测序。
[0075]
3.“种”水平的分类学鉴定使用获得的v3
‑
v4区16s rrna基因,使用常规方法进行质控,去除模糊碱基、嵌合体。然后使用肠道细菌16s rrna基因v3
‑
v4区参比序列工作库进行比对查询。将比对发现的一致性为100%的序列,按照比对上的参考序列的分类学信息,注释为已知细菌或未知细菌。如果注释为已知细菌,则使用相应的分类学名称注释,如猪链球菌。如果注释为未知细菌,使用相应编码的opu进行注释,包括疑似新种、高分阶单元等。不能注释的序列,注释为未知序列(unidentified)(图1)。
[0076]
4.人粪便菌群多样性和构成比的分析结果
[0077]
(1)本发明方法可从分类学“种”的水平,检测并描述人肠道菌群的多样性。本发明发现,每个健康中国人肠道菌群平均含有186
±
51个opu,其中低频菌群(10%以下人群携带)、中频菌群(10%
‑
60%以下人群携带)、高频菌群(60%以上人群携带)的opu数目分别为20
±
11、75
±
29和90
±
19。累计检出1235个opu,其中774个(62.7%)opu是未知细菌(图2
‑
图4)。
rrna基因全长测序,我们采用opu策略对每个样品的菌种组成和丰度信息分析。具体方法可以参考yang j,pu j,lu s,bai x,wu y,jin d,cheng y,zhang g,zhu w,luo x,rossell
ó‑
m
ó
ra r,xu j.species
‑
level analysis of human gut microbiota with metataxonomics.front microbiol.2020aug 26;11:2029.doi:10.3389/fmicb.2020.02029.pmid:32983030;pmcid:pmc7479098。
[0091]
3.分析的结果
[0092]
120个健康人样本根据测序方法.根据使用数据库和比对软件不同,共分为3种方法。分别为:(1)采用illumina miseq平台进行16s rrna基因v3
‑
v4区测序,采用本发明中构建的数据库和比对软件进行分析(以下简称为本发明方法);(2)采用llumina miseq平台进行16s rrna基因v3
‑
v4区测序,采用silva_132 16srrnadatabase数据库和rdp classifier贝叶斯算法进行比对分析(以下简称为常用方法);(3)采用pacbio sequel平台进行16s rrna基因全长测序,采用操作系统发生学单元策略对每个样品的菌种组成和丰度信息分析,因该方法能够获得16s rrna基因的全长序列,而采用16s rrna基因全长进行定“种”是金标准方法(以下简称为金标准方法)。根据分析结果,我们从能够确定到“种”级别序列条数比例和发现“种”的数量两个方面对三种方法进行比较分析,用于确定本发明中构建的数据库和比对软件具有优异的发现“种”的能力。
[0093]
(1)本发明方法能够将每份粪便标本平均95%以上的16s rrna基因序列鉴定到“种”的水平(opu)
[0094]
我们将本发明中建立的数据库和比对方法与16s rrna基因全长测序(金标准方法)进行比较,结果显示120个健康人样本中,本发明中方法鉴定到“种”水平的序列条数平均比例为95.6%(能够注释到opu的序列数/每份标本所有16s rrna基因v3
‑
v4区序列数)。而16s rrna基因全长测序(金标准方法),鉴定到“种”水平的序列条数平均比例为57.95%(能够注释到opu的序列数/每份标本所有16s rrna基因全长序列数)。以上数据说明本发明中的方法与金标准方法相比,在鉴定到“种”水平序列条数的比例提高方面更有优势。因金标准方法需要获得16s rrna全长序列,获得相同序列条数的情况下,测序成本约是本发明中方法的10倍以上,测序周期约是本发明中方法的2
‑
3倍,因此说明本发明中方法在确定“种”方面更具经济性和实用性。
[0095]
表2.粪便标本可鉴定到细菌“种”水平的16s rrna序列数(%)的比较*
[0096] 本发明方法金标准方法常用方法平均值95.6387931457.9538616537.86043291最小值79.0862737717.306076734.708447103最大值99.6310901995.2070851886.63499335中值97.2692567659.650477638.1376648
[0097]
*:本发明方法和常用方法(使用silva_132 16srrna database数据库加rdpclassifier贝叶斯算法):能够注释到opu的序列数/每份标本所有16s rrna基因v3
‑
v4区序列数;金标准方法:能够注释到opu的序列数/每份标本所有16srrna基因全长序列数。
[0098]
我们使用相同数据,即llumina miseq平台进行16s rrna基因v3
‑
v4区测序数据,分别采用本发明中构建的数据库加比对方法和目前常有的silva_13216srrnadatabase数据库加rdp classifier贝叶斯算法进行分析,并对确定到“种”级别序列条数进行对比。对
比结果显示,本发明中建立的数据库和比对方法平均能够将95.6%的序列鉴定到“种”水平,而目前常用的silva_132 16srrnadatabase数据库加rdp classifier贝叶斯算法只能将38.1%的序列鉴定到“种”水平。
[0099]
(2)本发明方法每份粪便标本能够检测到的细菌“种”数平均可达92.9(opu)
[0100]
表3、每个粪便标本能够检测的细菌“种”(opu)的数量比较
[0101] 金标准方法本发明方法常用方法平均值92.9137931140.474137982.07758621最小值349961最大值171179108中值94139.581
[0102]
在发现“种”的数量方面,本发明中构建的数据库和比对软件在120个样本中,平均每个样品发现140.47个“种”,而金标准方法平均每个样品中发现92.91个“种”,目前常用的数据库和比对软件(例如:silva_132 16srrna database数据库加rdp classifier贝叶斯算法)平均每个样本种只能发现82.08个“种”(见附表3)。以上数据说明本发明中构建的数据库和比对软件能够发现更多的“种”,对于肠道菌群结构和丰度分析具有重要的价值。
[0103]
应用实施例2:临床病人样本采用本发明方法进行粪便菌群组成及构成比分析
[0104]
我们利用分析120个健康人体肠道菌群16s rrna基因数据,确定健康人群中不同组成肠道菌的标准阈值,构建人体肠道菌群标准常规检查的参考标准。图5为检出率为60%及以上的、称之为肠道常驻菌群的116个opu的构成比阈值。在此基础上,我们针对3名临床患者的粪便标本,采用本发明方法进行了粪便菌群组成及丰度分析,并和参考人群菌群菌结构及丰度进行了对比分析,可为评估患者肠道菌群状况进行了分析,也涉及到和疾病的相关性。图5显示了健康人肠道菌群多样性和构成比的分析结果。
[0105]
人体肠道菌不仅能影响体重和消化能力、抵御感染和自体免疫疾病的患病风险,还能控制人体对疾病治疗药物的反应。因此,研究获得人体肠道菌群多样性和构成比数据,可作为健康、疾病状态的指示剂。医生通过解读人体菌群多样性和构成比数据,分析、判断、诊断患者的疾病和健康状况。
[0106]
应用实施例2.1:成人腹泻病粪便样本菌群分析
[0107]
病人编号f32,女,67岁,临床诊断为“志贺痢疾杆菌引起的细菌感染”。图6显示了腹泻患者肠道菌群多样性和构成比结果。从菌群结构及丰度结果可以看出得出如下结论:
[0108]
1、被检测粪便样本中escherichia coli/shigella丰度明显增高,显著高于阈值(0.6%)。
[0109]
2、检测出条件致病菌enterobacter asburiae,acinetobacter junii(健康人无检出)。
[0110]
3.合计发现细菌“种”数(opu)13个。健康人每份粪便标本可检测到opu140个(99
‑
179)。提示菌群多样性降低,菌群紊乱。
[0111]
4、由于16s rrna基因全长序列的一致性高于98.7%(一致性为98.7%及以上者可看作是一个“种”),仅仅依据16s rrna基因序列,无法将escherichia coli与shigella菌属分开。但escherichia coli/shigella丰度明显增高,支持志贺痢疾杆菌感染的临床诊断。
[0112]
应用实施例2.2:针对临床肝硬化病人样本2进行的分析
[0113]
病人编号f54,男,42岁,临床诊断为“肝硬化”。图7显示了患者肠道菌群多样性和构成比结果。从菌群结构及丰度结果可以看出得出如下结论:
[0114]
1、被检测粪便样本中bacteroides fragilis,klebsiella pneumoniae,ruminococcus torques等条件致病菌的丰度高于阈值。
[0115]
2、合计发现细菌“种”数(opu)69个。低于健康人平均每个粪便标本可发现140.47个“种”(99
‑
179opu)。提示菌群多样性降低,肠道菌群紊乱。应用实施例2.3:针对临床腹泻病人样本3进行的分析
[0116]
病人编号f181,男,1岁,临床诊断为“腹泻”,图8显示临床病人粪便标本群结构及丰度。从菌群结构及丰度结果可以看出得出如下结论:
[0117]
1.被检测粪便样本中,citrobacter braakii和citrobacter freundii丰度明显升高(阈值)。由于citrobacter braakii和citrobacter freundii可引起腹泻,可能是病原菌。
[0118]
2.klebsiella pneumoniae的丰度高于阈值。klebsiella pneumoniae可引起小儿腹泻。
[0119]
3.合计发现细菌“种”数(opu)52个。低于健康人平均每个粪便标本可发现140.47个“种”(99
‑
179opu)。提示肠道菌群多样性降低,肠道菌群紊乱。
[0120]
4.检测到益生菌lactobacillus reuteri和bifidobacterium breve,且丰度高于成年健康人数据。建议询问患者是否服用益生菌制剂。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1